
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Дипломная работа: Обследва процеса на реформинг на природния газ и получаване на водород
Дипломная работа: Обследва процеса на реформинг на природния газ и получаване на водород
Министерство на образованието и науката
Университет Професор доктор Асен Златаров”
Факултет Технически науки”
гр. Бургас
ДИПЛОМНА РАБОТА
на Петър Стефанов Делчев, ф. Н. ТНГ 4012
Специалност:
«Органични химични технологии. Технология на нефта и газа»
Научен ръководител:
доц. Т. Паличев
Дипломант:
инж. П. Делчев
І. Въведение
Водородът е най-простият, най-лекият и най-често срещан в природата елемент. Навсякъде е в свързано състояние – във водата, в сложните молекули на въглеводородите нефта, в природния газ. Има три изотопа: протий, деутерий и тритий, като най-разпространен е първият. Водородът изобилства в Слънчевата система като съставлява 71% от нейната маса или 91% от всички атоми.
Притеглен от гравитационните сили на Слънцето водородът се превръща в хелий чрез реакции на ядрен синтез, процесът който осигурява енергия за живот на Земята.
Молекулярният водород е газ без цвят и миризма и е практически неразтворим във вода. Противно на всички останали газове той се стопля при разширение, но само до определено налягане. Водородът е 14,5 пъти по-лек от въздуха и 16 пъти по-лек от кислорода и съгладно закона на Грем, той дифундира 4 пъти по-бълзо от кислорода във въздуха. Коефициентът му на топлопроводност е 7 пъти по-голяма от тази на въздуха.Водородът се втечнява при -253 0С, а изкристализира при -259 0С.
В химично отношение при обикновени температури водородът не е много активен. Енергично се свързва само с флуора, с хлора - фотохимично, а с кислорода при нагряване. При подходящи високи температури взаимодейства с много елементи като получените съединения се наричат хидриди.
Водородът е едно изключително перспективно гориво – при изгаряне отделя 120 МКал/кг, като продукт от изгарянето е само вода. В космическите совалки отдавна го използват за снабдяване с енергия, а получената вода астронавтите използват за пиене. Създаването на ДВГ работещ с водород би подобрило значително екоравновесието на земята.
Поради опасността от експлозия и неудобството при съхранение и транспорт, водородът все още не е намерил широко приложение като гориво за ДВГ. Най-използван метод на съхранение е в балони, като с увеличение на допустимото налягането, намаля масовото съдържание на водорода в съхранявания газ(300атм – 13% mass, 500атм – 11% mass, 900атм – 9% mass). Надежда за развитието на водорода като гориво е, че той се разтваря много добре в няко метали (600 до 3000 обема в паладий) и при загряване се освобождава. Друга насока в изследванията е използване на въглеродни нанотръбички или стъклени микросфери за съхраняване на водорода. От друга страна разтварянето на водорода в желязото е причина за намаляне на механичните му показатели (т. н. „водородна слабост”), което е причина за аварийни ситуации. В инсталации за производство на водород се използват скъпи, висококачествени стомани.
Съществуват следните методи за получаване на водород:
· Термохимични – наричат се още реформинг процеси и в момента намират най-широко приложение. Използват термокаталитично преобразуване на въглеводороди като природен газ, метанол, газолин и дори въглища във взаимодействие с водни пари.
· Електрохимични – при пропускане на електрически ток през воден разтвор на електролит се получава водород
· Фотоелектрохимични – водородът се получава, когато към потопен във вода електрод, през който протича ток, се насочи светлина.
· Фотобиологични - фотобиологичните системи принципно използват естествената фотосинтетична активност на бактерии и зелени водорасли, които синтезират водород.
В настоящето водородът е основна суровина, без който е немислимо съществуването на торовата промишленост, производството на горива, в хранителната промишленост за произходство на хидрирани мазнини, за получаване на някои метали(W, Mo) в чист вид от техните оксиди поради неговото редукционно действие.
Водородът в нефтохимическата промишленост се получава основно при процесите на реформинг и крекинг и се използва за хидроочистка на средно и високо дестилатните фракции от серни и азотни съединения. Без него е немислимо производството на дизелови горива. В хидрокрекинг процесите се използва за получаване на изключително висококачествени бензини. Инсталациите за реформинг са източник на водород, поради протичащите в тях процеси на дехидрогенизация и циклизация на въглеводородите, но някои нефтопреработвателни заводи строят специални инсталации за производство на водород.
В торовата промишленост водородът се използва за синтез на амоняк, от който се произвеждат едни от най-високотонажните минерални торове – амониева селитра и карбамид.
Цел на настоящата дипломна работа е да се обследва процеса на реформинг на природния газ и получаване на водород като съставна част на азото-водородния синтез газ в цех АМ – 76 на „Неохим” АД.
ІІ. Теоретична част
В днешни дни основен метод за производство на водород е паро-каталитическата конверсия на въглеводородите, в частност природения газ. Нефтопреработвателната и нефтохимическа индустрия разполага с значителни ресурси на водородсъдържащи газове, които обаче не могат да се използват, заради ниското съдържание на водород в тях и присъствието на вредни примеси. Този метод е добре разработен технологично и апаратурно.
За метода на паро-кислородната газификация нефтените остатъци важи същото, но при него са нужни по-големи експлуатационни разходи и капиталовложения. Що се отнася до другите методи за получаване на водород като основен продукт (крекинг на въглеводороди, метало-паров метод, електролиза на водата, термохимични методи), те не са намерили приложение в промишлеността и затова няма да бъдат разгледани в тази дипломна работа.
Водородът в процесите на частично дехидриране (каталитически риформинт на бензина, дехидриране на бутана и бутилена), се явава страничен макар и ценен продукт.
1. Паро-каталитична конверсия на въглеводородите
Производството на водород по този метод се състои в три етапа: подготовка на суровината, конверсия и отделяне на оксидите на въглерода от конвертирания газ. На стадия на подготовка суровината се очиства от ненаситени въглеводороди, органични съединения на сярата, в някои случаи се провежда стабилизация, посредством частична конверсия на хомолозите на метана. На стадия на отделянето на въглеродните оксиди от конвертирания газ се извършва конверсия на въглеродния оксид с водна пара, очистка на газа от въглероден диоксид и отделяне на остатъчните оксиди на въглерода чрез метаниране. Изброените етапи, с изключение на отделянето на въглеродния диоксид, са каталитични процеси близки по апаратурно оформление.
1.1 Подготовка на суровината за каталитична конверсия
Катализатоторите използвани при парова конверсия на въглеводородите, нискотемпературна конверсия на въглеродния оксид и метаниране лесно се отравят от серни съединения. В изходната суровина може да се съдържат като примеси сероводород, меркаптани, сулфиди, тиофен и др.
В процеса на парова конверсия на въглеводородите на никеловия катализатор съединенията на сярата се хидрират с образуване на сероводород. Той взаимодейства с никеловия катализатор по реакцията:
mNi + nH2S →NiimSn + nH2
Отравянето на катализаторите, използвани за нискотемпературна конверсия на въглеродния оксид става в резултат на неговото взаимодействие с сероводород по реакцията:
ZnO + H2S →ZnS + H2O
Cu + H2S → CuS + H2
В суровината трябва да отсъстват примеси на ненаситени въглеводороди, наличието на които създава опасност от отложения на кокс върху катализатора.
В качеството на суровина за парова конверсия се използват различни газове, но най-често метан. В нефтопреработването, използваните за процеса на конверсия газове са странични продукти с променлив състав. Затова е целесъобразно на стадия на подготовка да се осигури стабилен по състав газ, чрез нискотемпературна конверсия на хомолозите на метана в метан по реакцията:
CnH2n+2 + (n-1)/2H2O → (3n+1)/4CH4 + (n-1)/4CO2
1.1.1 Очистване от серните съединения и ненаситени въглеводороди
Изискванията към дълбочината на очистване от серни съединения за различните стадии на производство на водород са различни и зависят от условията, при които се води процеса и от използвания катализатор. Отравяне на никеловия катализатор се наблюдава още при съдържание на сяра 0,1% от масата на катализатора, въпреки че за покриване повърхността му е нужно около 1% сяра.
Равновесното съдържание на никелов сулфид при конверсия се снижава с увеличаване на температурата. За различните катализатори пределно допустимата концентрация на H2S в газа в зависимост от съдържанието на никел и активността на катализатора е (в мг/м3):
при 800 0С – от 1 до 60
при 850 0С – от 5 до 76
при 900 0С – от 25 до 120
Твърде чувствителен към отравяне със сероводород са катализаторите за нискотемпературна конверсия на въглеродния оксид, съдържащи медни и цинкови оксиди. Например при съдържание на сяра в газа 0,2 мг/м3 и обемна скорост 3000 ч-1 срокът на използване на катализатора НТК-4 е две години. Отчитайки увеличения обем на газа в процеса с 4-6 пъти, концентрацията на сените съединения в очистения газ, постъпващ за конверсия, се ограничава до 1 – 1,5 мг/м3. Още по-големи са изискванията към дълбочината на очистване на газа за нискотемпературната конверсия на метана. Съдържанието на сероводород в очистения газ в този случай не трябва да превишава 0,5 мг/м3.
В условията на парова конверсия, ненаситените въглеводороди образуват циклични структури и полимеризират с образуване на кокс, което води до блокиране на активните центрове на катализатора. Едновременно с това протичат реакции на хидрогениране и парова конверсия на ненаситени въглеводороди:
CnH2n + 2H2 → CnH2n+2
CnH2n + nH2O → nCO + 3nH2
Съдържанието на ненаситени въглеводороди в суровината се ограничава от съотношението на скоростта на тези реакции и скоростта на образуване на кокс.
Много серни съединения се разлагат термически при нагряване до 400 0С. Продустите от терморазпада на серните съединения са сероводород и съответните олефини, но процесът е съпроводен с образуването на високомолекулни смолисти вещества. Например меркаптаните и дисулфидите се разлагат при 200 0С, но тиофена и сулфидите не се разлагат и при 400 0С. Ако в суровината има само термически неустойчиви серни съединения, очистването се провежда в един стадий с поглътител на основата на цинковия оксид. С негова помощ от газа се отделя сероводорода, серовъглерод и меркаптани, но не се отделят тиофена и сулфидите.
По-надеждна е двустепенната схема за извличане на серните съединения из въглеводородната суровина, вчключваща деструктивно хидриране на серните съединения и последващо поглъщане на сероводорода с цинков оксид.
В интервала 300 – 400 0С, в които се осъществява процеса на очистване, реакцията на хидриране на серните съединения е практически необратима.
Концентрацията на водорода в нефтозаводските газове, постъпващи за хидриране е 10 -15%. При хидриране на серни съединения, съдържащи се в природния газ или бензина, към суровината се добавят още 5 – 7% водород. Концентрацията на органаничните серни съединения не превишава 50 мг/м3, тоест 103 – 104 пъти по-ниска от тази на водорода.
В инсталациите за парова конверсия на природен газ или нефтозаводски газове, работещи при ниско налягане, със съдържание на ненаситени въглеводороди не повече от 0,5%, за очистване на серните съединения се използват контактни маси 481-Cu и 481-Zn. Процесът се води при 300-350 0С. Тези поглътители се зареждат в тарелкови реактори по слоеве, при което първият се използва за преобразуване на серните съединения в сероводород, а втората – за поглъщане на H2S. Следва да се отбележи, че и 481-Cu в значителна степен реагира и със сероводорода като го поглъща, а цинковият оксид поглъща и такива съединения като меркаптаните, серовъглерод и други. Необходимата дълбочина на очистване се достига при обемна скорост до 1000 м-1 като се отчете и общия обем на контактните маси.
При повишено налягане (2 – 3 MPa) в стадия на хидриране се използват модифицирани алумокобалтомолибденов и алумоникелмолибденов катализатор, намиращи приложение за хидрогенизационни процеси в нефтопреработката и нефтохимията.
Между серните съединения върху катализатора и сярата и водорода, намиращи се в газа се установява равновесие. При промяна на съдържанието на сяра или водород в газа равновесието се нарушава и е възможно отделяне на сяра от катализатора или поглъщане на сяра от газа. При очистването по двустепенната схема този процес обаче не дава отражение на общия ефект от очистката, доколкото след хидриращия катализатор следва поглътител на серните съединения на основата на цинкови оксиди. Взаимодействието на сероводорода и цинковия оксид при 350 – 400 0С и излишък на водород протича до край.
На основата на цинка, освен 481-Zn, се изработват поглътители ГИАП-10 и ГИАП-10-2. За очистване от сероводород е възможно използването също на отработен катализатор за нискотемпературна конверсия на въглеродния оксид НТК-4., съдържащ цинков оксид. Стандартният цинков оксид има малка относителна повърхност (4,2 – 6,6 м2/гр) и много ниска сярополгътимост (1,7 4,2%). Неговото използване като очистващ реагент е нецелесъобразно. Активираната форма на цинковия оксид се получава при разлагането на цинков карбонат или хидрооксид при 350 – 400 0С. При това се получава цинков оксид с относителна повърхност 32,8 м2/гр и серопоглътимост 32%, а при разлагане на цинков хидрооксид – относителна повърхност 26,9 м2/гр и серопоглъщаемост 21,9%. ГИАП-10 се получава на основа цинков карбонат, а 481-Zn – цинков хидрооксид. Въвеждането на меден оксид в катализатора позвалява реакционната температура да се намали до 260 -280 0С.
В табл. 1 са приведени характеристиките на някои катализатори с хириращо действие, а в табл. 2 на катализатори-поглътители на серните съединения на основата на цинков оксид.
Табл. 1
М-8-10 Al-Co-Mo Al-Ni-Mo
Размер на частиците, мм
диаметър.................. 2,8 - 3,0 4,0 4,1 4,0 – 4,5
височина................... 4 8 5,0 – 5,5 4,0 – 4,5
Относителна плътност,
кг/м3.................................. 690 640 640 - 670
Активна повърхност,
м2/гр................................. 242 150 100 - 120
Активност*, % mass......... 99 98,4 95
* Активността характеризира дълобочината на превръщане на
C2H5SH, съдържащ се в природния газ, при 350 0С и обемна
Скорост 2000 ч-1 при количество на добавяния водород 5%
Табл. 2
| Поглъти-тел | Произход (фирма, страна) |
Форма и размер на частиците,(мм) |
Относи-телна плътност, кг/м3 |
Пори- стост, % |
Активна повърхност, м2/гр |
Серопоглъ-щаемост при 400 0С |
|
ГИАП-10 ГИАП-10-2 B-ZnO G-72C 32-4 29-2 R-5-10 |
6-03-322-72 6-03-328-72 BASF(Герм) Gerdler(САЩ) ICI(Англия) ICI(Англия) ICI(Англия) |
Таблетки d = 4 -5 h = 3 – 4 Същото Гранули d=4 h=5-10 Сфери r=2-3 Сфери r=3 Таблетки d=5 h=5 Таблетки d=4 h=4-6 |
1400 – 1500 2110 1400 1230 1190 1930 1600 |
45 -55 36,9 44,8 58,6 43,3 30,3 - |
32,8 45,0 18,7 35,6 34,9 35,7 20,0 |
25,0 19,6 26,3 21,4 20,2 20,7 22,0 |
В схемата на инсталации, предвиждащи провеждане на конверсия на въглеводороди при 2,2 – 2,4 MPa, на стадия на очистка на серните съединения е целесъобразно да се използва алумоникелмолибденосиликатен катализатор и поглътител ГИАП-10. Тогава и за двата стадия условията на очистка са еднакви: температура 350 – 400 0С, обемна скорост 1000 ч-1, налягане 2,3-2,5 MPa. Такива условия са благоприятни за хидриране на ненаситените въглеводороди, които се превръщат в съответните наситени въглеводороди по реакция:
CnH2n + H2 → CnH2n+2 + Q
Термодинамически в дадения температурен интервал е възможно също да протичат реакции на хидрокрекинг на наситените въглеводороди с образуване на метан:
CnH2n+2 + (n - 1)H2 →n CH4 + Q
В таблица 3 са дадени константите на равновесие и топлинните ефекти на реакцията на хидриране на ненаситените въглеводороди и хидрокрекинг на етан и пропан.
Както се вижда от таблицата, хидрирането на ненаситените въглеводороди в интервала 300 – 400 0С практически протича до край. Такива термодинамично благоприятни условия има и за хидрокрекинг на наситените въглеводороди, но хидрокрекинг на наситените въглеводоро-ди C2 – C5 не протича на катализатори съдържащи сяра и при наличие на серни съединения в газа.
Табл. 3
|
Темпера- тура, 0С |
Реакция на хидриране |
Реакции на хид- ро крекинг |
||||
|
C2H4 |
C3H6 |
н-C4H8 |
изо-C4H8 |
C2H6 |
C3H8 |
|
|
200 300 400 500 600 700 800 900 1000 |
5,1.108 9,8.105 8,3.104 1,1.104 3,9.102 3,1.10 4,1 7,8.10-1 6,1.10-2 |
1,4.106 2,8.104 3,5.103 6,8.102 2,5.10 2,3 3,7.10-1 9,1.10-2 1,0.10-2 |
9,2.106 3,2.104 4,1.103 8,0.102 3,2.10 2,8 4,3.10-1 1,0.10--1 1,0.10-2 |
1,0.106 5,1.103 6,5.102 1,1.102 6,7 7,9.10-1 1,4.10-1 4,1.10-2 6,3.10-3 |
-- 1,5.106 2,03.105 4,47.104 1,33.104 4,97.103 2,22.103 -- -- |
-- 4,62.1011 1,04.1010 5,90.108 5,98.107 9,15.106 1,97.106 -- -- |
| Q, kJ/mol | 137 | 126 | 127 | 119 | 68 | 127 |
Провеждани са опити за едновременно очистване на нефтозаводските газове от серни съединения и хидриране на ненаситените въглево-дороди в лабораторни условия с използване на алумокобалтомолибденов и алумоникелмолибденов катализатор и поглътител ГИАП-10. Условията и резултатите от опита са следните:
Продължителност на изпитанието, ч 3000
Налягане, MPa.................................... 2,0
Температура, 0С................................. 380 - 400
Обемна скорост, ч-1............................ 1000 – 1500
Съдържание на H2 в суровината, % 35 60
Съдържание на ненавитени въгле-
Водороди, %
в суровината............................... 5 – 20
в изходящите газове.................. отсъства
Съдържание на меркаптани, мг/нм3
в суровината.............................. 5,3 – 28
в изходящите газове................. 0,1 – 0,9 0,1 – 0,3
(на Al–Co–Mo) (на Al-Ni-Mo)
Лабораторните опити погазали, че хидриране на ненаситените въглеводороди протича до край, в същото време хидрокрекинг на наситените въглеводороди не се наблюдава.
Реакцията на хидрокрекинг на серните съединения е екзотермична, но впредвид малките количества на тези съединения в суровината, топлинния ефект може да бъде пренебрегнат. При значително съдържание на ненаситени съединения в суровината, в резултат на тяхното хидрира-не, се повишава температурата на процеса. Например, при съдържание на 2,5% етилен, температурата на газа на изход от реактора се повишава с 25 0С. В такъв случай, ако се хидрира газ, съдържащ повече от 4% ненаситени въглеводороди, за да се избегне прегряване на катализатора е необходимо да се обезпечи отвеждане на топлината от реактора.
Продължителността на работа на катализатора, използван за очистване на суровината от серни съединения и ненаситени въглеводороди, обикновено е 2 – 3 години. Срокът на използване на поглътителя зависи от съдържанието на H2S в газа. Ако сяроуловяемостта на поглътителя е недостатъчна или концентрацията на серни съединения е висока, в схемата се предвижда включване на два последователно действащи апарата с поглътител.
Нефтозаводските газове след моноетаноламиновата очистка от сероводород съдържат 50 – 100 мг/м3 серни съединения. В този случай про серопоглъщаемост 20% от масата, срокът на използвана за едно зареждане с поглътител е 3 – 6 хил. часа, т.е. доста по-малко от срока на използване на хидриращите катализатори. Затова е нерационално да се използват така наречените бифункционални катализатори, представляващи полгътител с добавка от хидриращ агент и предназначени за гидрогенолиза на серните съединения и едновременно поглъщане на сяроводо-рода. Освен това, на бифункционалните катализатори хидрирането на ненаситените въглеводороди не протича с достатъчна дълбочина.
1.1.2 Частична конверсия на хомолозите на метана
Паровата конверсия на въглеводороди с преимуществено образуване на метан, наричаща се частична конверсия, в днешно време намира приложение за получаване на заместители на природния газ, от въглеводороди достигащи до С8 – С10.
Пълна конверсия на нефтозаводските газове и бензини в тръбни реактори с външно подгряване фактически протича в два стадия: първи – частична конверсия – парова конверсия на хомолозите на метана преи-муществено в метан в началния участък на реакционната зона и втори – конверсия на метана и получаване на водород и въглеродни оксиди. Първият стадий може да се осъществи в отделен реактор при 350 – 500 0С, в режим призък до адиабатния. Това би позволило по-ефективно да се използват скъпите пещи за конверсия с тръбни реактори за провеждане на основната реакция за пълна конверсия на метана и да се съкрати разхода на пара, без опасност от отложение на кокс върху катализатора.
Частичната конверсия може да се разглежда като стадии за подготовка, чрез стабилизация на суровината, за пълна парова конверсия на метана. В този случай, налягането на процеса на частична конверсия се определя от налягането на вход в тръбния реактор за пълна конверсия. Отношението пара/газ не може да бъде по-високо от това в стадия на пълна конверсия, но то може да се намали до 2/1 с цел да не се натоварва реактора за частична конверсия с излишно количество пара.
В процеса на частична конверсия протичат реакции с отделяне на топлина (хидрокрекинг на въглеводородите с образуване на метан) и с поглъщане на топлина (парова конверсия на метана, служеща като донор на водород за реакциите на хидрокрекинг). Колкото е по-висока температурата, толкова по-силно развитие получава процеса на конверсия на метана. Температурата на процеса на частична конверсия се избира така, че процесът да протича при адиабатни условия. Реактора за частична конверсия е разположен след реактора за очистване на газа от серни съединения, където температурата на процеса е 350 – 400 0С. Поради това, долната граница на процеса на частична конверсия е 350 0С, а гор-ната се определя от условията на адиабатност – 500 0С. В табл. 4 са представени данни за термодинамическите характеристики по състава и добива на газ при частична конверсия на нефтозаводските газове при 2,4 MPa, и 400 0С с различен въглероден еквивалент на изходящия газ (n) приведен в табл. 4
Табл. 4
|
Въглеро- ден екви- валент |
Разход на пара, м3/м3 |
Състав на газа, %(об.) |
Добов на газ, м3/м3 |
Количество отделяща се топлина, kJ/m3 |
||||
|
CO2 |
CO |
H2 |
CH4 |
сух | влажен | |||
|
1,0 1,2 1,4 1,6 1,8 2,0 2,2 2,4 2,6 2,8 3,0 |
2,0 2,4 2,8 3,2 3,6 4,0 4,4 4,8 5,2 5,6 6,0 |
4,95 7,69 9,77 11,41 12,73 13,81 14,72 15,50 16,16 16,74 17,25 |
0,06 0,07 0,08 0,09 0,09 0,09 0,09 0,08 0,08 0,08 0,07 |
19,95 17,18 15,08 13,43 12,12 11,03 10,13 9,36 8,70 8,12 7,62 |
75,04 75,06 75,06 75,07 75,06 75,06 75,06 75,06 75,06 75,06 75,06 |
1,24 1,44 1,64 1,84 2,04 2,24 2,44 2,64 2,84 3,04 3,24 |
3,12 3,62 4,12 4,62 5,12 5,62 6,12 6,62 7,12 7,62 8,12 |
422,8 542,0 663,5 784,9 904,2 1025,8 1147,2 1268,0 1385,8 1507,2 1628,7 |
1.2 Процес на паро-каталитична конверсия на въглеводо-родите
1.2.1 Термодинамични основи на процеса
На съвременните катализатори конверсията на въглеводородите протича при условия доближаващи се до термодинамическото равновесие. Реакциите на парова конверсия са обратими. Състава и добива на продуктите се определя от термодинамическото равновесие на протичащите реакции.
Конверсията на метана с водна пара може да се опише с реакциите:
CH4
+ H2O ![]() CO + 3H2
206,4 kJ (1)
CO + 3H2
206,4 kJ (1)
CO + H2O ![]() CO2 + H2 + 41,0 kJ (2)
CO2 + H2 + 41,0 kJ (2)
Константите на равновесие на реакциите (K1 и K2) се изразяват с уравненията:

където PCO, PH2, PCO2, PH2O, PCH4 – парциалните налягания на съответстващите компоненти в равновесната смес.
Началният и крайният състав на конвертирания газ, а също парциалните налягания на неговите компоненти при установяване на равновесие се изразяват с уравненията, приведени в табл. 5.
Табл. 5 Уравнения на равновесния състав на конвертирания газ и парциалните налянагия на неговите компоненти при конвесия на метана
| Компоненти |
Обем на газовата смес, м3 |
Парциално налягане на компонентите в конвертирания газ, MPa |
|
| Начален | След конверсията | ||
|
CH4
H2O CO CO2 H2 |
1 a - - - |
1 – α a – α – β α – β 3α + β |
(1 – α).Р 1 + a +2α (a – α – β).P 1 + a + 2α (α – β).P 1 + a + 2α β.P 1 + a + 2α (3α + β).P 1 + a + 2α |
| Общо | 1 + a | 1 + a + 2α = V | P |
Легенда: a – обем на подадената пара за единица от обема CH4; α – част на реагиралия CH4; β – част на реагиралия CO; Р – налягане в систе-мата; V – общ обем на конвертирания газ.
Замествайки съответно на парциалните налягания на компонентите в уравненията на константите на равновесие на реакциите се получава:
K1= [(α – β).(3α + β)3].P2
(1 α).(a – α – β).(1 + a + 2a)2 (5)
K2= β.(3α + β)
(α – β).(a – α – β) (6)
Хомолозите на метана C2 – C10 са по-неустойчиви при еднакви температурни условия отколкото метана, което е видно от данните за константите на равновесие на реакциите на конверсия на метана, етана и пропана с водна пара, приведени в табл. 6. Термодинамичните разчети са показали, че при 300 0С и по-висши хомолози на метана присъвстват в конвертирания газ в нищожни количества.
Табл. 6 Константи на равновесие на реакцията на кон версия на метана, етана и пропана в водна пара
|
Температура, 0С |
KCH4 |
KC2H6 |
KC3H8 |
|
327 427 527 627 727 827 |
5,058.10-7 2,687.10-4 3,120.10-2 1,306 26,56 3,133.102 |
3,805.10-7 1,467.10-2 43,281 2,268.104 3,505.106 2,184.108 |
5,686.10-8 0,2015 1,775.104 1,331.108 1,716.1011 6,084.1013 |
Термодинамичните изчисления на паровата конверсия на алифатните въглеводороди, започваща от 300 0С, може да бъде водена използвайки уравненията за парова конверсия на метана (2) и окиси на въглеро-да (3), а също и уравнението за парова конверсия на хомолозите на метана в метан (1). Предвид последните уравнения за стехиометричните съотношения на компонентите и техните парциални налягания при паровата конверсия на алифатните въглеводороди приемат значенията, приведени в табл.7.
Табл. 7 Уравнение за равновесния състав на конвертирания газ и парциалните налягания на неговите компоненти при конверсия на алифатните въглеводороди
| Компо-ненти |
Обем на газовата смес, м3 |
Парциално налягане на компонентите, MPa |
|
| Начален | След конверия | ||
|
CnH2n+2 CH4 H2O CO CO2 H2 |
1 - (n-1)/2 + [(3n + 1)a]/4 - - - |
- (3n + 1)/4.(1 α) (3n + 1)/4.(a – α β) (3n + 1)/4.(α – β) (3n + 1)/4.β + (n 1)/4 (3n + 1)/4.(3α + β) |
- (3n + 1)/4.(1 α).P/V (3n + 1)/4.(a – α β).P/V (3n + 1)/4.(α β).P/V [(3n + 1)/4.β + (n 1)/4].P/V (3n + 1)/4.(3α + β).P/V |
| Общо |
1 + (n – 1)/2 + + (3n + 1)/4.a |
(n – 1)/4 + (3n + 1)/4. .(1 + a + 2α) = V |
P |
Замествайки парциялните налягания със значенията им от таблицата, в уравненията за константите на равновисие ще получим
K1 = (α – β).(3α + β)3. [(3n – 1)/4.P/V]2
(1 α).(a – α – β) (7)
K2 = [(n – 1)/(3n +1) + β](3α + β)
(α – β).(a – α – β) (8)
Уравнения (5) и (6) са частен случай на уравнения (7) и (8), когато n=1.
Последните уравнения позволяват да се изчисляват състава и добива на конвертиран газ не само за процеса на пълна конверсия на въглеводородите, когато преобладаващи компонент стават H2, CO, CO2, а съдържанието на CH4 неголямо, но и за частична конверсия на въглеводородите, когато преобладаващ продукт на реакциите се явява CH4.
С помощта на горните уравнения и таблици с метода за последователното приближение се извършват дигитални изчисления за условията на протичане на реакциите на парова конверсия за съвременните схеми за производство на водород. Във връзка с това, че в реални условия рав-новесието на реакциите на конверсия на метана с пара не се достига, за практически изчисления на K1 се приема температура, по-ниска от темпе-ратурата на изход от реактора. При провеждане на процеса под налягане 2 – 3MPa разликата е 20 -30 0С.
Паровата конверсия на въглеводородите се провежда така, че на катализатора да не се отлага водород. Закоксуването с въглерод върху катализатора може да доведе до неговото разрушение и увеличаване на съпротивлението на катализаторния слой. Затова едновременно с описаните по-горе изчисления се правят и разчети на термодинамичното равновесие на реакциите с възможно образуване на въглерод в системата по една от следващите реакции:
CnH2n+2
![]() nC +(n +
1)H2 – Q
nC +(n +
1)H2 – Q
CO
+ H2 ![]() C + H2O + Q
C + H2O + Q
2CO ![]() C + CO2 + Q
C + CO2 + Q
Константите на равновесие на реакциите на разпадане на въглеводородите приведени в приложение в табл. 8. Константите на равновесие на последните две реакции K3 и K4 са следните:
Температура K3 = PCOPH2 K4 = (PCO)2
PH2O PCO2
300 1,43.10-5 3,60.10-7
500 2,01.10-2 4,51.10-3
700 2,00 1,32
900 2,97.101 3,90.101
1100 2,25.102 4,72.102
Табл. 8 Константи на равновесие на реакциите на разпадане на въглеводородие
|
Тем- пера- тура, 0С |
KCH4 |
KC2H6 |
KC3H8 |
KC2H4 |
KC3H6 |
KC2H2 |
|
200 400 600 800 1000 1200 1400 |
9,3.103 2,0.10 6,9.10-1 8,5.10-2 2,0.10-2 7,1.10-3 3,2.10-3 |
1,4.10 3,3.10-3 3,3.10-5 1,9.10-6 2,6.10-7 6,3.10-8 2,1.10-8 |
32.10-2 1,0.10-6 1,5.10-9 1,1.10-10 9,8.10-12 1,7.10-12 4,7.10-13 |
5,5.10-8 3,5.10-7 9,1.10-7 1,7.10-6 2,5.10-6 3,5.10-6 4,4.10-6 |
3,2.10-10 2,5.10-10 2,2.10-10 2,0.10-10 1,9.10-10 1,8.10-10 1,7.10-10 |
2,5.10-23 1,4.10-14 1,7.10-11 6,3.10-9 3,6.10-7 6,9.10-6 6,6.10-5 |
За
определяне на термодинамичните условия за отложение на въглерод по реакция CO + H2 ![]() C + H2O намираме
константата на равновесие по формулата:
C + H2O намираме
константата на равновесие по формулата:
K3 = PCOPH2
PH2O
Използвайки парциалните налягания на реакцията на конверсия от табл. 7 ще получим за K3 следното уравнение:
K3 = (α – β)(3α + β)(1 + a + 2α).V
(a α – β) P
Минималният разход на пара в процеса на парова конверсия на нефтени газове и бензини, под който е възможно закоксуване, се определя като се решат съвместно последното уравнение с уравнения (7) и (8). Тогава ще получим:
K3 = (α – β)(3α + β)(3n + 1).V
(a – α – β) P
Тези уравнения обаче не се отнасят за конверсия на газове, съдържащи ненаситени и ароматни въглеводороди, които на вход в реакторната зона могат да се разпаднат с отделяне на въглерод.
Процесът на парова конверсия се извършва с топлообмен през стените на реактора. Количеството на подаваната топлинна енергия може да се изчисли след като са определени добива и състава на конвертирания газ. Сметките се правят възоснова на първия закон на термодинамиката по уравнението на топлинния баланс:
q = Qн’ + t’.(c’ + ac’) – V.(Qн’’ + c’’t’’)
, където q топлината подадена отвън в процеса на парова конверсия (ПК) на 1 м3 изходящ газ, kJ/m3; Qн’ – топлината на изгаряне на изходния газ, kJ/m3; Qн’’ – топлината на изгаряне на влажния конвертиран газ, kJ/m3; t’ и t’’ – температура на парогазовата смес на вход в реактора и температура на конвертирания газ на изход от реактора, 0С; c’ – средна топлоемкост на изходния газ и водната пара при постоянно налягане и температура на вход в реактора, kJ/(m3.0C); c’’ – средна топлоемкост на влажния конвертиран газ при постоянно налягане и температура на изход от реактора, kJ/(m3.0C); V – обем на влажния конвертиран газ, получен за 1 м3 от изходния газ, м3; a – обем на водната пара, подавана за 1 м3 от изходния газ, м3.
1.2.2 Режим на процеса
Режимът на паро-каталитичната конверсия на въглеводородите трябва да осигурява получаване на технически водород със съдържание 95 – 98% H2. При по-ниска концентрация се повишават разходите на инсталацията за хидрокрекинг. Производството на водород с висока концентрация изисква големи капита-ловложения и не е оправдано икономически. Технически водород с концентра-ция около 95% водород може да се получи при съдържание в сухия конвертиран газ 2 – 2,5% метан, тъй като в следващите процеси на очистване от CO2 след конверсията на въглеродния оксид и метаниране съдържанието на метана в газа нараства до 4 – 5%. Степента на конверсия на метана в такъв случай е 0,9. Технически водород със съдържание 98% Н2 се получава при съдържание в конверти-рания газ на 1 – 1,3% СН4 или степен на конверсия на метана 0,95.
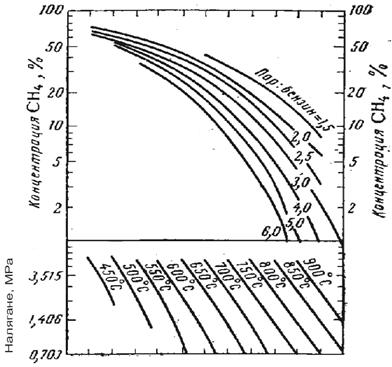
Водород със съответната концентрация може да бъде получен чрез изменение на налягането, температурата и отношението пара/метан. Връзката между тези параметри е илюстрирана на фигура 1 и 2.
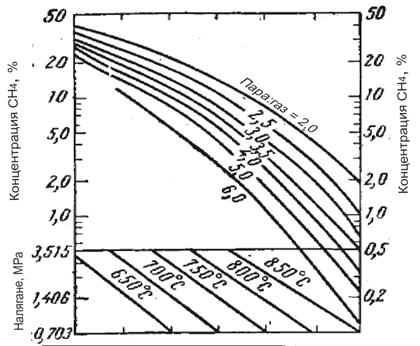
Фиг. 1 Зависимост на конц. СН4 в сухия газ от температурата, на негането, отношението пара/метан в условията на достигнато термодинамично равновесие
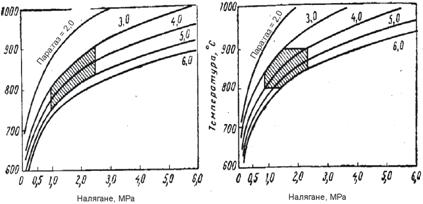
Фиг.2 Режими на процеса на ПК на метана, осигуряващи в условията на ТД равновесие получаване на 95% Н2 и 96% Н2 (съответно първата и втора графика; защрихованите об ласти са условията, при които се работи в съществуващи те инсталации)
Както се вижда от фигурите, режимът може да се променя в широки диапазони, но техническите възможности на оборудването, а също и режимите на другите стадии на производство и отделянето на въглерод при определени гранични условия, значително стесняват този диапазон. Резултатите от пресмятането за минимален разход на пара, под които се отделя въглерод, са показани във фи-гура 3.
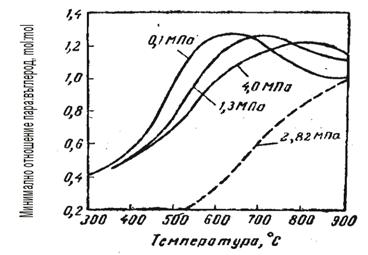
Фиг. 3 Граници на отделяне на въглерод в процеса на ПК при различни налягания (с непрекъсната – ПК на бензин, прекъсната – ПК на метан)
Разхода на пара за конверсия на метана трябва да е не по-нисък от 2:1, за да се предотврати отделянето на въглерод, но такова отношение не са използва, защото в случай парата трябва да се добавя на стадия на парова конверсия на въглероднидния оксид. В реактора за ПК за излишното количество добавена пара се изразходва допълнителна топлина, но то се възстановява в котел-утилизатора. Подаване на излишък от пара подобрява топлопредаването, затова за 1м3 метан се изразходва не по малко от 3 м3 водна пара, а при налягане 2 МРа са не-обходими 4 – 5 м3.
Режима на ПК е ограничен не само от разхода на пара, но и от температу-рата, налягането. Максималната температура на процеса зависи от качеството на стоманата, диаметъра на реактора, допустимото топлинно напрежение на върху повърхността на реакционните тръби и особено от налягането на процеса. В голяма част от инсталациите температурата на процеса се поддържа в границите 830 – 880 0С. При по-ниска температура е трудно да се получи водород с необходимите качества, а под 750 0С процесът е неефективен. В интервала 750 – 800 0С ПК се осъществява при ниско налягане, но провеждане на процеса при наля-гане под 1,0 МРа е нецелесъобразно.
Изборът на налягане е обусловен най-вече от възможността да се използва топлината от кондензация на нереагиралата пара за регенерация на разтвора в системата за очистване на газа от СО2. При налягане по-ниско от 0,7 – 1,0 МРа, използването на тази топлина е практически невъзможно. Освен това, повишение на налягането се прави с цел понижаване на енергиините разходи от ком-пресиране на газа. Комресирането на суровината, в сравнение с компресирането на водорода, позволява да се съкратят енергиините разходи, пропорционално с увеличение обема на газа в процеса на ПК. Повишеното налягане интензифици-ра масообмена и топлопренасянето в реакторите и топлообменниците. Обикновено ПК на метана се води при налягане 1,2 – 3,0 МРа, без да се взема под внимание, че повишеното налягане премества равновесието на реакцията на ПК към неблагоприятните зони (фиг. 1 и 2).
За достигане на необходимата концентрация на водород при увеличено на-лягане, се увеличава температурата на процеса и разхода на пара. Но повишено-то налягане и температура водят до нуждата от използване на реакционни тръби от високолегирани стомани. Във връзка с това производството на водород в дне-шни дни се осъществява при налягане на по-високо от 2,5 МРа. Границата на во-дене на процеса е обусловена от качеството стоманата на реакционните тръби, данни на фигура 4. В съвременните инсталации, процеса се води при 2,0 – 2,6 МРа, 800 880 0С и отношение пара:метан от (4 – 5) : 1.
Ограниченията в съдържанието на Н2, температурата и налягането при избора на режема на ПК на метана важат и за конверсията на нефто-заводските газове и бензини.
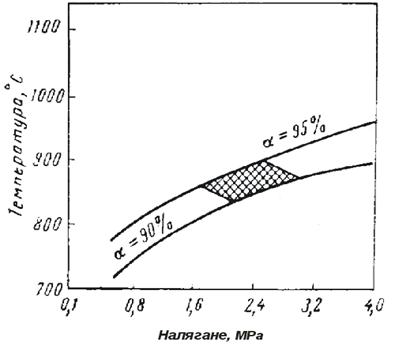
Фиг. 4 Ограничение на режима на паро-каталитична конверсия от качеството на стоманата на реакционните тръби
В таблица 9 и 10 са приведени резултатите от ТД изчисления за ПК на нефтозаводски газове с различен въглероден еквивалент n за производство на 95% и 98% водород при 2 МРа и температура 830 и 860 0С (температурата на конвертирания газ на изход от реактора ше е съот-ветно 800 и 830 0С)
Табл. 9 Добив и състав на конвертирания газ от ПК на нефтозаводските газове в условия на достигнато ТД равновесие при 2 МРа и 830 0С
| n |
Разход на пара, м3/м3 |
Добив на газ, м3/м3 |
Състав на сухия газ,% | ||||
| сух | влажен |
СО2 |
СО |
Н2 |
СН4 |
||
|
Съдържание на Н2 в технически водород 95 – 96% |
|||||||
|
1,0 1,4 1,8 2,2 2,6 3,0 |
4,5 6,1 7,6 9,2 10,7 12,2 |
4,1 5,4 6,7 8,0 9,3 1,5 |
7,3 9,6 11,9 14,1 16,4 18,6 |
10,6 11,6 12,1 12,5 12,8 13,0 |
11,1 11,7 12,2 12,5 12,7 12,9 |
75,7 74,1 73,1 72,4 71,9 71,5 |
2,6 2,6 2,6 2,6 2,6 2,6 |
|
Съдържание на Н2 в технически водород 98% |
|||||||
|
1,0 1,4 1,8 2,2 2,6 3,0 |
6,0 8,2 10,2 12,2 14,2 16,2 |
4,3 5,7 7,1 8,5 9,8 11,2 |
8,9 11,8 14,6 17,4 20,1 22,9 |
12,1 13,1 13,7 14,1 14,4 14,6 |
9,6 10,0 10,3 10,6 10,8 10,9 |
77,0 75,6 74,1 74,0 73,5 73,2 |
1,3 1,3 1,3 1,3 1,3 1,3 |
Както се вижда в таблица 10 с увеличаване на въглеродния еквива-лент на нефтозаводските газове, расте и разхода на пара за 1 м3 от изходходния газ, но относителния разход на пара расте незначително. По-забележимо (24 – 25%) расте разхода на пара при повишение концен-трацията на Н2 в техническия водород от 95 на 98%.
Табл. 10 Добив и състав на конвертирания газ от ПК на нефтозаводските газове в условията на достигнато ТД равновесие при 2 МРа и 860 0С
| n |
Разход на пара, м3/м3 |
Добив на газ, м3/м3 |
Състав на сухия газ, % | |||||||
| сух | влажен |
СО2 |
СО |
Н2 |
СН4 |
|||||
|
Съдържание на Н2 в технически водород 95 – 96% |
||||||||||
|
1,0 1,4 1,8 2,2 2,6 3,0 |
3,6 4,9 6,2 7,4 8,7 9,9 |
4,0 5,3 6,5 7,8 9,1 10,3 |
6,4 8,4 10,4 12,4 14,4 16,4 |
8,8 9,6 10,1 10,5 10,7 10,9 |
13,3 14,7 14,7 15,1 15,3 15,5 |
75,3 73,6 72,6 71,9 71,4 71,0 |
2,6 2,6 2,6 2,6 2,6 2,6 |
|||
|
Съдържание на Н2 в технически водород 98% |
||||||||||
|
1,0 1,4 1,8 2,2 2,6 3,0 |
4,9 6,6 8,2 9,9 11,6 13,2 |
4,3 5,6 6,9 8,3 9,7 11,0 |
7,8 10,2 12,6 15,1 17,6 19,9 |
10,5 11,4 11,9 12,2 12,6 12,7 |
11,6 12,2 12,7 13,0 13,1 13,3 |
76,6 75,1 74,1 73,5 73,0 72,7 |
1,3 1,3 1,3 1,3 1,3 1,3 |
|||
С увеличение на въглеродния еквивалент на газа, относителния разход на пара спрямо въглерода, по данни от ТД изчисления, малко се понижава. На практика обаче, поради опасност от отделяне на въглерод на катализатора (особено при нестабилен състав на газа), с увеличавене на въглеродния еквивалент на газа, разхода на пара се повишава. По-високият въглероден еквивалент n на нефтозаводските газове, в сравнение с природния, по-малката им стабилност, водят до необходимост от увеличен разход на пара. Зависимостта на разхода на пара от n на суровината е дадена на фигура 5.
На фигура 6 е дадена зависимостта на равновесната концентрация на метана в сухия газ при конверсия на бензина от температура, налягане и отношение пара:бензин(номограмата може да служи за приб-лизителна оценка на режима на процеса).
Общите зависимости на процеса от температурата и налягането за хомолозите на метана са същите като тези за самия метан.
Изборът на температура и налягане на процеса на ПК на нефтоза-водските газове, а също и бензини се определя от същите фактори, които се определя избора при конверсия на природен газ.
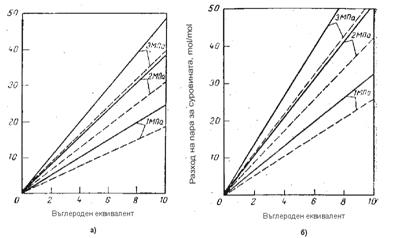
Фиг. 5 Зависимост на разхода на пара от въглеродния еквивалент при ПК на въглеводороди и производство на 95 – 96% Н2 а) и 98% Н2 б) при температура 830 0С (непре късната линия) и 860 0С (прекъсната)

Във връзка с разработването на устойчиви паладиеви мембрани е предложена инсталация за конверсия на метан с отделяне на водорода от реакционната зона през мембрана. Това би преместило ТД равновесие на реакцията. Изчисленията за ТД равновесие на реакцията на ПК при налягане 1,925 МРа, отношение пара:метан равно на 3:1 и парциално налягане на Н2 в остатъчния газ 0,16 МРа, показали, че при отвеждане на получения Н2, още при 500 0С степента на конверсия на метана достига 1, като в същото време без отвеждане на Н2 степента на конверсия на СН4 0,9 може да се достигне едва при 880 0С.
Количеството чист водород, което минава през мембраната, се оп-ределя също от парциалното налягане на Н2 в остатъчния газ, на изход от реактора. На фигура 7 е показан резултата от ТД изчисления на ПК на метана с извеждане на водород при различни температури и парциални налягания на Н2 в остатъчния газ. Както следва от приведените данни, воденето на процеса при 600 – 700 0С е по-ефективно, тъй като се пови-шава количеството на водорода, извеждан през мемраната. С увелича-ване на температурата възниква възможност за повишаване на парциал-ното налягане Н2 в остатъчния газ, което интензифицира дифузията на водорода през мембраната.
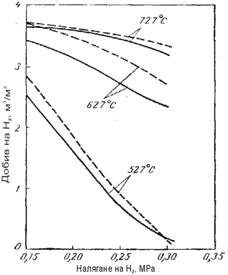

Показателите на процеса, които могат да се достигнат при едновре-менно отвеждане на водород от реакционната зона са следните:
Налягане, МРа...................................................... 2,0
Температура, 0С................................................... 727
Отношение пара:метан........................................ 2
Парциално налягане на Н2 в остатъчния
газ, МРа................................................................. 0,3
Температура на нагряване на паро-газовата
смес, 0С.................................................................. 430
Добив, м3/м3
водород 100%-ен............................................. 3,23
остатъчен газ................................................... 1,65
Състав на остатъчния газ, %
Н2...................................................................... 15,2
СО..................................................................... 17,5
СО2.................................................................... 39,6
СН4.................................................................... 3,5
Н2О.................................................................... 24,2
Степен на конверсия на СН4................................ 0,94
Степен на извличане на Н2.................................. 0,93
1.2.3 Катализатори за парова конверсия на въглеводо-родите
Паровата конверсия на метана без катализатор протича с приемли-ва скорост и дълбочина на превръщане върху пълнеж от активни глини само при температури 1250 – 1350 0С. Опитите осъществени в кух кварц-ов реактор, показали, че при обемна скорост 200 ч-1, отношение пара:газ равно на 2:1 и атмосферно налягане, даже при 1000 0С степента на кон-версия на метана не надвишава 8 – 9%, а при 900 0С тя е равна на 1,1%. При нагряване на хомолози на метана в смес с водни пари, без катализа-тор, над 500 – 600 0С протичат с висока скорост реакции на пиролиз с об-разуване на ненаситени въглеводороди (етилен, пропилен и др.). Образу-ват се още метан, етан, пропан и малко водород.
Катализаторите за ПК на въглеводородите са предназначени не само за ускоряване на основната реакция, но и за инхибиране на страни-чните реакции на пиролиз. Крайно нежелателни странични реакции са тези, при които се отделя въглерод. На тези реакции противостои газифи-кацията на въглерода с водни пари.
Паровата конверсия на въглеводородите се осъществява в стома-нени реакционни тръби (d = 100 – 150 мм) с външно подгряване. Паро-газовия поток се движи през слой от катализатор с височина до 12 м. Максимално възможната обемна скорост на суровината зависи не само от активността на катализатора, но и от скоростта на подаване на топли-на, а също и от съпротивлението на катализаторния слой.
В резултат на разработването на високоактивни катализатори, лимитиращ фактор при провеждане на процеса се е оказало подаването на топлина. Ускоряването на топлопреноса в слоя катализатор при прове-ждане на процес под налягане, а също и усъвършенстване конструкцията на пещите и увеличаване дължината на реакционните тръби довело довело до това, че съпротивлението на слоя катализатор станало лими-тиращ фактор на процеса. То в значителна степен зависи от размера и формата на частиците на катализатора и за понижаване съпротивление-то на слоя се използват катализатори с по-големи размери и с пръстено-видна форма.
Процесът на конверсия протича на активната повърхност на катали-затора. Външната повърхност на частиците е относително неголяма и тя се увеличава за сметка на увеличение пористостта на катализатора и създаване на развита вътрешна повърхност.
Към катализатора има изисквания относно неговата термична, хи-мична и механична устойчивост. Срокът на използване на катализатора не трябва да е по-малък от 2 години. Катализатора за ПК на въглеводоро-дите се оценява по съвкупност от свойства като: активност, селективност, коефициент на хидравлично съпротивление, здравина и т.н.
Изброените изисквания не са изчертателни. В случай на временно нарушение в системата за очистване от серни съединения е възможно отравяне на катализатора, така че, той трябва да притежава способност за възстановяване на активността. При понижаване на отношениеот пара кам суровина, е възможно на катализатора да се отдели въглерод, което обикновено води до неговото унищожаване. Катализатор, който не се е разрушил, трябва да може да възстанови своята активност след газифи-кация на въглерода. Също, катализаторът не трябва да отделя летливи компоненти като серни съединения, силициеви оксиди и кисилини под действие на водните пари. Тези съединения може да попречат на по-нататъшното протичане на процеса.
Катализаторите за ПК съдържат активен компонент, промотор и но-сител. Като активен компонент се използва никел. Кобалтът е по-малко достъпен и затова не се използва. Паладий, платина, родий и рутений, въпреки, че са по-ефективни от никела са значително по-скъпи. Процеса се катализира от активните центрове, затова основно значение има акти-вната повърхност, а не общата повърхност.
Носителят придава на катализатора механична здравина, позваля-ва да се съкрати разхода на активния компонент и да обезпечи необходи-мата високоразвита повърхност. Никелът на повърхността на носителя е във вид на малки кристали. В условията на парова конверсия при високо съдържание на пара и температура по-висока от 750 0С е възможно нара-стване на кристалите и свързано с това понижаване на активността. Носителят трябва да възпрепятства този процес, т. е. трябва да стабили-зира катализатора и не трябва да променя свойствата си до температура до 900 0С. В качество на носител се използва алуминиев оксид в α – мо-дификация, оксиди на магнезия и оксиди на силиция.
Неголемите добавки от промотор (1 – 1,5%) към основния компо-нент позволява да се повиши и съхрани активността на катализатора. Като промотор се използват оксиди на магнезия и алуминия. В някои ка-тализатори с цел интензификация на реакцията на газификация на въгле-рода, в неголеми количества се добавят алкални метали. Във връзка с летливостта на алкалите са предложени катализатори на основа оксиди на урана.
Използват се два метода за производство на катализатори. По пър-вият соли на металите се смесват с прахообразен носител и се формоват чрез екструзия от мокра тестообразна маса или чрез сухо пресоване. Често в качество на свързваща добавка се използва цимент.
По втория метод, върху формувания и закален носител, чрез дву-, три- или четирикратно напояване с воден разтвор на соли се нанася на-пример Ni(NO3) върху α-Al2O3. По втория метод се получават катализатори съдържащи до 10% Ni. Катализатор с по-високо съдържание на никел се получава чрез формуване. За по-добро смесване на компонентите, те биват съвместно утаявани под формата на хидрати, оксиди или карбона-ти.
Готовият катализатор, независимо от метода на получаване, се из-пича при температура 400 500 0С, при което солите на Ni преминават в кристална форма.
С особено внимание трябва да се добавя към носителя магнезиев оксид, тък като в резултат на неговата хидратация е възможно да се раз-руши катализатора или да се намали здравината му. Хидратацията може да стане при понижаване на температурата под 425 0С, затова при темпе-ратури по-ниски от 500 0С е задължително да се избягва контакт на ката-лизатор съдържащ MgO с водна пара. Калциевият оксид, в състава на носителя, ако не е химически свързан, напримес с алуминиев оксид(кал-циев алуминат), също е способен на хидратация.
При конверсия на нефтозаводски газове и бензини има опасност от отлагане на въглерод на катализатора. За конверсия на бензини, фирма ICI е разработила катализатор 46-1 с добавка от акален метал. В този катализа-тор калият е химически свързан с алумосиликати, образувайки комплекс-ни съединения, например KAlSiO4. В условията на ПК под действие на водна пара и въглероден диоксид тези съединения бавно се разлагат с образуване на неголеми количества калиев карбонат. Той предотвратява отлагане на въглерод, но от друга страна е летлив и постепенно се унася от реактора. Влошава се работата на котел-утилизатора, поради отложенията от калиев карбонат. За да се избегне това е предложено на изход от реактора да се зарежда друг катализатор 46-2, който химически свързва калия. Последният катализатор едновременно с това и служи за конверсия на метана.
Магнезиевият оксид също способства за предотвратяване отделяне на въглерод върху катализатора. Има съобщения за разработка на ката-лизатор с високи якостни показатели от магнезиев оксид без добавка от алкални метали, съдържащ 7% Ni. Вследствие някои свойства на магне-зия не се отделя въглерод дори при отношение пара:газ = 2:1.
Като най-радикално решение на проблема с конверсия на хомоло-зите на метана следва да признаем двустепенния процес на ПК. В 1-ва степен процеса се води в адиабатен реактор при 450 – 520 0С с получава-не на газ, съдържащ преимуществено метан. Във 2-ра степен се провеж-да пълна конверсия на метана в реакционните тръби с външно нагряване и с използване на добре представящи се катализатори. В последните години за частична конверсия на въглеводородите са разработени високоефективни и устойчиви катализатори.
Катализаторите за частична конверсия също съдържат никел като активен компонент. По-ниската температура на процеса изисква използ-ване на по-активни катализатори. Повишаване на активността на катали-затора се постига чрез увеличаване на концентрацията на никел до 40 – 60%, увеличаване на относителната повърхност до 100 – 300 м2/гр и относителната повърхност на активния компонент никел до 20 – 60 м2/гр. Следва да се отбележи, че повишаване концентрацията на никел в катализатора не означава увеличаване на неговата относителна повърх-ност, което е видно от следните данни:
Концентрация на никел, %........ 5 17 30 53 73
Относителна повърхност на
активния компонент Ni, м2/гр.... 1 14 25 52 43
Активността на катализатора зависи от носителя и метода на про-изводството му. За катализатор за частична конверсия се използват малки сферички от алуминиев оксид γ–модификация. По-доброто раз-пределение на никела се постига при утаяване на никел съвместно с алу-миний в разтвор на тези метали. Падащите при това аморфни сфери от хидрогел, при по-нататъшна кристализация се разпадат на по-малки кристали, образувайки голяма контактна повърхност. В качество на про-мотор може да се използва магнезиев оксид, титанов диоксид и циркони-ев оксид.
Табл. 11 Характеристика на катализаторите за конверсия на въглеводороди, произвеждани в Русия
| Показатели | ГИАП-3 | ГИАП-3-6н | ГИАП-5 | ГИАП-16 | ГИАП-15 |
|
Химичен състав, % NiO ……………………. Al2O3 ………………….. CaO …………………... MgO …………………... SiO2 …………………… Fe2O3 …………………. TiO2 …………………… Гранула-пръстен с размер, мм Гранула-пръстен с размер, мм Гранула-цилиндър с размер, мм Относителна плътност, кг/м3 Относителна повърхност, м2/гр Пористост, %............................ Средна механична якост, МN/m2 |
4,5 95,0 - - - - - 11х11х3 15х15х5 20х20х7 8х9 1400 8 33 – 39 60 - 90 |
7,0 86,0 - - - - - 8х9 12х12 1650 8 33 90 |
23 – 26 24 – 30 10 – 15 11 – 17 18 – 25 ≤4 ≤2 10х10х4 14х14х6 19х19х8 - 800 – 900 30 – 50 31 50 |
25 46 14 15 - - - 15х17х11 - 1100 44 33 70 |
6 – 10 88 – 92 - - - - ≤2 12х12х6 - 1200 0,5 25 55 |
За да се предотврати отделяне на въглерод към катализатора се добавят алкални метали, катализиращи газификацията на въглерода. В този случай добавката на алкален метал е по-оправдана, отколкото при каталицаторите за пълна конверсия, доколкото до 500 0С алкалите почти не се унасят. От алкалните и алкалоземни метали, най-често се препо-ръчва добавка на калий, макар че има и препоръки за Ba, Mg и Са. Изпол-зват се също катализатори съдържащи 10 – 30% Ni на Al2O3. В такъв ка-тализатор се добавя 5 – 25% Zn или Cr и се промотира с вече споменати-те добавки.
Основните производители на катализатори са фирми като Gendler (САЩ), ICI(Англия), BASF(Германия). Фирма Gendler произвежда катали-затор G-56 с различно съдържание на никел в пръстеновидна форма. Фирма ICI има катализатор 57-1, предназначен за ПК на природен газ и нефтозавод-ски газове, несъдържащи ненаситени въглеводороди, при налягане до 3 МРа и температура до 850 0С. Съставът на катализаторите от фирма ICI е следния:
марка............ 57-1 46-1 марка........... 57,1 46-1
Ni . 32 21 Al2O3 ……….. 54 32
CaO . 14 11 MgO ………… - 13
SiO2 . 0,1 16 K2O …………. - 7
1.2.4 Загуба на активност на катализатора
Под действие на каталитичните отрови, катализаторите могат час-тично или изцяло да загубят своята активност. В редица случаи е възмо-жно тя да се възстанови, ако не нацяло, то поне частично, след прекратя-ване действието на отровителя. Някои вещества отрявят катализатора необратимо. Към каталитическите отрови следва да се отнесат: сярово-дород и органични съединения на сярата, съединения на арсена, халоге-ните, фосфора, оловото и медта. Суровина и пара, подадени отделно или в недостатъчни количества също могат да са отровители.
Загуба на активността е вследствие химичното взаимодействие на никела с катализаторния отровител и образяване на неактивни съедине-ния. За отравяне на катализатора е нужно в такова взаимодействие да са встъпили дори само активните центрове на Ni. Например, катализатор съдържащ 15% Ni бил отровен при 750 0С, когато съдържанието на сяра в него било едва 0,005%, т. е. когато само 0,069% от Ni са образували сул-фид. По-активните катализатори, по принцип са и по-чувствителни към отровители. Така повишаване качеството на катализаторите е довело до изискване към по-добро очистване на суровината от сяра.
Отравяне на катализатора със сяра е възможно при нарушаване режима на работа на инсталацията за сероочистка. При непродължител-ни нарушения, катализаторът в срок от няколко денонощия възстановява активността си. При отравяне на катализатора се нарушава кинетичното равновесие на реакцията на образуване и газификация на въглерода, което може да доведе до отлагане на въглерод и разрушаване на катали-затора.
Хлорът е силен отровител на катализаторите за ПК на въглеводо-родите и в още по-голяма степен за катализаторите за нискотемператур-ната конверсия на въглеродните оксиди. Йоните на хлора могат да попа-днат на катализатора от водните пари при недобро обезсоляване на химически очистената вода.
Катализаторът губи своята активност и при продължително въздей-ствие на водна пара, вследствие окисление на никела. В условия на ПК съотношението между окислителя (водни пари) и възстановяващия ком-понент (СО и Н2) е такова, че реакцията на възстановяване преобладава над реакцията на окисление и никелът в по-голяма част се намира във възстановено състояние. На вход в реактора при 400 500 0С конверсия не протича и възстановяването е слабо и в участък с дължина 1 - 3 м, къ-дето става подгряването на компонентите до 600 – 700 0С, процесите на окисление преобладават над тези на възстановяване. Така в началния участък никела се окислява и катализаторът губи своята активност, което от своя страна задържа началото на реакция. В такива условия началния участък работи като подгряващ за суровина и пара, което не е ефективно. Ако в газа постъпващ за конверсия има водород, условията за възстано-въване на катализатора в началните участъци се подобряват и никелът се съхранява във възстановен вид.
Окисление на никела в целия обем на катализатора може да стане при прекратяване или намаляване подаването на суровина и запазване подаването на пара. При 700 – 800 0С никеловият оксид взаимодейства с алуминиевия оксид, преобладаващ в носителя, с образуване на никелов алуминат (шпинел) по реакцията:
NiO + Al2O3 → NiiAl2O4
Полученият шпинел не притежава каталитична активност. Реакция-та протича по-бързо с γ-Al2O3 и значително по-бавно с α- Al2O3. Катали-затор съдържащ α- Al2O3 може да бъде нагряван с водни пари до 800 0С без да се образуват шпинели. Възстановяването на катализатор, в който никелът се е окислил не е сложно, ако пък са се образували шпинели, е необходимо третиране с водород при 800 0С.
1.2.5 Механизъм и кинетика на парова конверсия на въглеводороди
Механизмът на реакция между метан и водна пара на никелов ката-лизатор не е точно установен. Редица автори считат, че това е реакция от първи порядък, т. е. нейната скорост е право пропорционална на кон-центрацията на метана. Изследвания, направени върху реален катализа-тор, показали, че макрокинетиката на ПК на метана се подчинява на вътренодифузионния режим и скоростта на реакция (w, mol/s на 1м3 от катализатора) се описва с уравнението:
w = η.k.pCH4
където η-фактор на ефективноста, отчитащ масообмена; k – реална скоростна константа на реакцията (при отсъствие на вътрешнодифузинни пречки); pCH4 – парциално налягане на СН4, МРа.
Факторът на ефективност зависи от отношението между размера на катализаторните гранули и дълбочината на проникване на реакцията в тяхната вътрешност. По данни, дълбочина на проникване във въкрешно-стта на катализатор с диаметър 1,2 мм е само 0,04 мм. За катализатора на фирма Gerdler – G-56В, факторът на ефективност зависи от размера на частиците. При размер 6,4 мм η=0,216, а при гранули с размер 0,51 – 0,84 мм η=0,954. По тези данни дълбочината на проникване във вътреш-ността на гранулите е 0,2 – 0,3 мм. Тази величина свидетелства за това, че катализаторът работи с малка част от повърхностния слой. Така се оказва, че не повече от 5 – 8% от никела, съдържащ се в катализатора контактира с реагиращите вещества.
Намерена е примерна зависимост на скоростта на реакция от външ-ната повърхност на катализатора, която е свързана с малката дълбочина на проникване на реакцията във вътрешността на гранулите. Ако се вклю-чи фактора на ефективност в скоростната константа, уравнението ще приеме вида:
w = k.pCH4
За катализатор ГИАП-3, k се определя от уравнение:
k = 2,1.104.S.e-19400/RT
където S e относителната повърхност на катализатора, см2/гр.
В редица разработки са открити затормозяващи влияния от продук-тите на реакцията върху скоростта на ПК на метана. Затова, скоростта на реакция се описва с уравненията:
w = k. pCH4
pH2
w = k. pCH4.pH2O
10.pH2 + pH2O
w = k. pCH4
1 + a(pH2O/pH2) + b.pCO
Още по-малка е яснотата по отшение механизма на ПК на висшите въглеводороди. Установено е само, че в процеса на ПК хомолозите на метана се превръщат в метан, т. е. протича процес на частична конвер-сия. Предполага се, че въглеводородът, попаднал на повърхността на катализатора се дисоциира и образува радикали СНх, които реагират с водна пара и водород. В резултан на взаимодействие на радикали с вод-ни молекули, се образуват въглероден оксид и водород, а при взаимодей-ствие с водород – метан и въглерод. Така разрледан механизмът на кон-версия включва крекинг на въглеводороди, хидриране на продукти на кре-кинга и газификация на получения въглерод.
Има и други предположения за механизма на процеса на частична конверсия с преимуществено получаване на метан. Отначало става кон-версия на въглеводорода:

2СО + 2Н2 → СН4 + СО2
СО2 + 4Н2 → СН4 + 2Н2О
Противоположно мнение, предложено от други изследователи, че в началото се образува метан, който в последствие се подлага на конвер-сия с пара и се образува СО, СО2 и Н2. В други трудове е посочено, че първата реакция е на хидрокрекинг на въглеводородите:
СnH2n+2 + (n – 1)H2 → nCH4
, а след това е вече реакция на конверсия на метана.
Липсата на яснота около механизма на процеса не позволява да се даде кинетично уравнение за частичната конверсия. В своя труд L. U. Hyman се опитва да заобиколи това затруднение като предлага такава су-марна реакция за ПК на въглеводородите:
CnH2n+2 + mH2O → xCH4 + yCO2 + zCO + iH2
и математически задоволително описва резултатите от експеримента.
1.3 Парова конверсия на въглеродния оксид
Газът, получев в процеса на ПК и паро-кислородна газификация, съдържа заедно с водорода и метан, въглероден оксид и диоксид. Кон-центрацията на въглеродните оксиди в газа, получени при конверсия на различни въглеводородни суровини се колебае от 6 – 15%, а в газ, полу-чен от газификация на мазут достига до 45%.
В резултана на конверсия на въглеродните оксиди с водна пара се получава допълнително количество водород, равно на съдържащия се в газа СО. Реакцията протича без промяна на общия обем от реагенти, съ-провожда се от отделяне на топлина и не зависи от налягането. С пони-жаване на температурата, равоновесието се премества към образуване на водород и въглероден диоксид.
В реални условия на протичана на процеса на ПК на въглеводороди и паро-кислородна газификация на мазута, когато реакционната темпера-тура подмине 800 0С, пректически се достига равновесие на реакцията на конверсия на въглеродния оксид и концентрацията му в газа, постъпващ за конверсиям; тази концентрация обикновено отговаря на равновесната за максимана температура на конверсия на въглеводороди или газифика-ция. В отделни случаи се наблюдава по-ниско съдържание на СО, което може да се случи, ако реакция протича при по-ниска температура в колек-торите след изхода на газа от тръбите на пеща за конверсия на въглево-дороди или на агрегата за газификация. Този ефект е незначителен, по-ради краткото време на престой на газа в системата пред реактора за конверсия на СО.
Знаейки състава на газа постъпващ за конверсия на СО, и констан-тата на равновесие (таблица 12), може чрез уравнението за константите на реакцията и данните от материалния баланс да се пресметне равно-весната концентрация на СО и други компоненти. В таблица 13 са приве-дени данните за материалния баланс на конверсия на въглеродния оксид (изчислено за 100 мола от изходния газ).
Като заместим стойностите на равновесните концентрации на ком-понентите във влажния конвертиран газ в уравнението за константите на конверсия на СО с пара и го решим за „х”, ще пулучим
![]()
Табл.
12 Константи на равновесие а реакцията СО + Н2О ![]() СО2 + Н2
СО2 + Н2
|
Температура, 0С |
К1 |
Температура, 0С |
К1 |
|
200 250 300 350 400 450 500 520 540 560 580 600 620 640 660 680 700 710 720 730 740 750 760 |
2,279.102 8,651.10 3,922.10 2,034.10 1,170.10 7,311 4,878 4,215 3,670 3,220 2,843 2,527 2,259 2,031 1,835 1,666 1,519 1,453 1,391 1,333 1,279 1,228 1,180 |
770 780 790 800 810 820 830 840 850 860 870 880 890 900 910 920 930 940 950 960 970 980 990 |
1,135 1,092 1,053 1,015 9,793.10-1 9,457.10-1 9,139.10-1 8,837.10-1 8,552.10-1 8,282.10-1 8,025.10-1 7,781.10-1 7,549.10-1 7,328.10-1 7,118.10-1 6,918.10-1 6,728.10-1 6,546.10-1 6,372.10-1 6,206.10-1 6,047.10-1 5,896.10-1 5,750.10-1 |
Табл. 13 Материален баланс за изчисляване на равновисието на конверия на въглероден оксид
| Компоненти | Изходящ газ, mol |
Реагирало коли- чество, mol |
Полу-чено,mol |
Конвертиран газ, mol |
|
CO H2 CO2 CH4 |
a b c 1 – (a + b + c) |
x - - - |
- x x - |
a – x b + x c + x 1 – (a + b + c) |
| Общ сух газ | 1 | х | 2х | 1 + х |
|
Н2О |
е | х | - | е - х |
| Общ влажен газ | 1 + е | 2х | 2х | 1 + е |
Изчисленията показват, че за достигане на дастатъчно ниска кон-центрация на въглероден оксид в конвертирания газ, конверсията трябва да се провежда при температура не по-висока от 250 0С. С цел повишава-не производителността на процеса, конверсията обикновено се провежда на две степени: на първа се използва високотемпературен желязохромен катализатор, на втора нискотемпературен катализатор, на който медта е активен компонент. Преди откриването на нискотемпературните катали-затори, конверсията на въглероден оксид се е провеждала на железохро-мен катализатор при 350 – 450 0С в няколко степени. Високата степен на превръщане на СО се достигала за сметка на големия излишък на водна пара и очистване на конвертирания газ от въглероден диоксид между сте-пените. Такава схема е тромава и неикономична, затова с основно значе-ние на изследванията за този стадий е откриването на нискотемперату-рен катализатор.
Двустепенната конверсия на въглероден оксид на първи стадий се провежда при 350 – 400 0С, а на втори – при 220 – 250 0С. В газа, на вход на първа степен, отношението пара:газ се определя от режима на пред-ходния стадий (конверсия на въглеводородите) и трябва да се поддържа не подържа не по-ниско от 0,6:1 при налягане 2МРа и не по-ниско от 0,9:1 при налягане близко до атмосферното. Ако в газа получен на предходния стадий парата е недостатъчно, тя се добавя. Излишъкът на пара е благо-приятен за провеждане на конверсия, но при определени условия той мо-же да способства за отравяне на катализатора.
Обемната скорост също зависи от налягането, при което се осъще-ствява прощеса и се променя от 1000ч-1 при налягане, близко до атмос-ферното, до 2500ч-1 при 2 МРа. Налягането на процеса обикновено зави-си от налягането на предходния стадий. На инсталации за ПК на въглево-дороди, конверсия на въглеродния оксид се води при налягане близко до атмосферното или при 2,0 – 2,5 МРа. На инсталации за газификазия на-лягането може да бъде повишено до 15 МРа. Въпреки, че изменение на налягането не влияе на равновесието на реакцията, повишеното наляга-не се оказва благоприятно влияние на кинетичните фактори, които позво-ляват да се увеличи обемната скорост на газа.
В описаните условия конверсия на въглероден оксид протича с дъл-боко превръщане, близко до равновесното. Във всеки случай съдържа-нието на СО в газа след високотемпературна конверсия е 3 – 4%, а след нискотемпературна конверсия – до 0,5%. В реални условия дълбочината на превръщане на СО на стадия на високотемпературна конверсия е92 – 95%, а на стадия на нискотемпературна конверсия 90 – 92% от равно-весната дълбочина а превръщане.
За стадия на високотемпературна конверсия в началото се е из-ползвал катализатор на основата на Fe3O4. След това той е бил осъвър-шенстван с въвеждане на оксиди на хрома, възпрепятстващи увелича-ването на кристалите на Fe3O4, което увеличава срока на работа на ката-лизатора. Днес за този стадий на процеса се използват само модифика-ции на железохромния катализатор, отличаващи се по начина на произ-водство. Основни характеристики на двата железохромни катализатори № 482 и С-12-1 са следните:
№482 С-12-1
Форма.................................. гранули таблетки
Диаметър, мм..................... 4,5 – 5,5 9,7
Височина, мм...................... 5 – 20 4,9
Съдържание, %
желязо......................... 89 90 85,8
хром............................. 7,0 7,5 9,4
Активност* по скоростна кон-
станта при 350 0С, см3/гр ката-
лизатор в час..................... 1,0 – 1,3 1,30
Механична якост на
надлъжно напрежение, MN/m2 35 – 40 35
напречно напрежение, MN/m2 82,2 251
Относителна плътност, кг/м3 1150 1200
Относителна повърхност, м2/гр 25 – 30 110
Обем на порите, см3/гр..... 0,32 0,24
в това число с радиус
r < 7,5 нм 0,03 0,12
r > 7,5 нм 0,29 0,12
* активността е измерене на инсталация от проточен тип:
размер на зърната 1 – 2 мм, температура 350 0С, отно-
шение пара:газ = 3:1, обемна скорост 1200 ч-1
Срокът на използване на катализатора се определя основно от ме-ханичната якост; в процеса на работа той постепенно се разрушава, кое-то води до увеличаване на хидравличното съпротивление на слоя. Последното се наблюдава по-малко при използване на таблетирани ката-лизатори, но при тяхното използване се влошава дифузията и мате-риалът от вътрешността на таблетката се използва слабо. За подобрява-не на условията на дифузия се използва катализатор със ситно зърно или таблетки с малки размери. За намаляване на съпротивлението се използ-ват конвертори от радиален тип.
Желязохромният катализатор е малко чувствителен към отравяне от серни съединения, но съдържащите се в него или погълнати серни съединения, при взаимодействие с водород образуват H2S, който може да предизвика отравяне на катализатора в нискотемпературния стадий на конверсия. Затова при извеждане на инсталацията в режим газ от ре-актора, зареденият железохромен катализатор се изважда от схемата до-като в него се съдържа сероводород.
В състава на катализатора за нискотемпературна конверсия се въ-веждат мед, цинк и алуминий. В невъзстановена форма катализаторът е неактивен. В процеса на възстановяване CuO преминава в проста мед, която се явява катализатор на процеса. Цинковиятоксид изпълнява роля на стабилизатор, препятстващ увеличаване на размерите на кристала на медта, което може да намали активната повърхност на катализатора. За тази цел служи и алуминиевия оксид, а също и хромния оксид.
Активността и стабилността на работа на катализатора в голяма степен зависи от метода на производството му. Така например, цинко-медните катализатори с добавка на хромен оксид се приготвят чрез съв-местно утаяване имат висока активност, но не са достатъчно стабилни. Друг метод се състой в смесването на прахове от меден оксид и цинков оксид с концентриран разтвор на (NH4)2Cr2O7 и последващо закаляване и формуване. Катализаторите приготвени по този метод поддържат актив-ност на достатъчни високо ниво в продължение на 1 – 2 години. Такъв е срокът на работа на цинкомедните катализатори на фирмите Gendler, CCI и ICI. Активен и стабилно работещ катализатор ГИАП, се приготвя чрез разтваряне на Сu2CO3 в хромен анхидрид (CrO3) и смесвана разтвора със суспензия на цинков оксид. Получената маса се промотира, суши, закаля-ва и таблетира. В качество на промотор се използва манганов оксид, алу-миний, магнезий и титан.
Нискотемпературният катализатор е много чувствителен към отра-вяне от серни съединения и халогени. Условията на процеса са благопри-ятни за образуване на цинков и меден сулфид, но механизмът на отравя-не е свързан преди всичко с образуване на цинков сулфид и възникващо-то от това уголемяване на медните кристали. Аналогично е действието на йоните на хлора. Отравянето става послойно по ход на газа. Рязкото понижаване активността на катализатора в долните части на слоя се наб-людава при съдържание на сяра от 0,12%. Концентрацията на хлора, дос-татъчна за отравяне на катализатора е под аналитично измеримото ниво (0,1 мг/м3). За да се избегне отравяне на катализаторите, във връхната зона,първ по хода на конвертирания газ, се зарежда поглътител предста-вляващ смес от цинков оксид и активен алуминиев оксид. Използват се също и зеолити.
Друга причина за дезактивация на катализатора може да бъде прег-ряване на слоя, предизвикващо неговото спичане. За високотемператур-ния катализатор не трябва да се допускат температури по-високи от 500 0С, за нискотемпературния – по-високи от 260 0С. Опасност от прегряване може да възникне както при възстановяване, така и в процес на конвер-сия, доколкото реакциите на възстановяване на железния и медния оксид и реакцията на конверсия протичат с отделяне на значително количество топлина.
Количеството топлина отделящо се на стадия на високотемпера-турна конверсия, зависи преди всичко от концентрацията на СО в конвер-тирания газ. В адиабатен реактор повишението на температурата на кон-вертируемия газ в реални е около 10 0С на всеки процент превърнат въг-лероден оксид. Обикновено съдържанието на въглероден оксид в газа, получен от паровата конверсия на въглеводородите не превишава 6% от-несено към влажния газ. В такъв случай високотемпературната конверсия на СО може да се проведе в адиабатен реактор в една степен. При по-ви-соко съдържание на въглероден оксид процесът се провежда в няколко степени с междинно охлаждане на конвертирания газ.
Газът постъпващ за нискотемпературна конверсия, независимо от състава на изходната суровина, съдържа не повече от 2,5% СО. Следо-вателно повишението на температурата не трябва да е повече от 25 0С, което е напълно допустимо, а прегряване е допустимо само при наруше-ние в режима на работа на предходния стадий. Характеристиката на изменение на температурата в катализаторния слой е свързана с разпре-делението по дължината на реакционната зона на реакцията на въглеродния оксид и зависи от активността на катализатора (фигура 8)


Относно кинетиката на конверсията на въглеродния оксид е предло-жен механизъм на реакция на повърхността на оксидния катализатор:
Н2О + () →Н2 + (О)
СО + (О) →С О2 + ()
Н2О + СО →С О + Н2
, където (О) – атомът на кислорода на повърхността на катализатора, () – активния центърна повърхността на катализатора.
За реакции, протичащи в кинетичната област е предложено уравне-нието:
w = k.pH2O.pCO – k-1.pCO2.pH2
B.pH2O + pCO2
, където w е скорост на реакция в mol/s на 1 м3 от катализатора; k – кон-станта на скоростта, В – отношение на скоростта на правата и обратна ре-акция.
Зависимостта на k и В от температурата за железомолибденовия катализатор е следната:
k = - 34000 + 10,2 B = 8800 + 2,32
4,57. T 4,57.T
За по-точно отределяне на кинетичните фактори, J. S. Cambell и В. И. Атрощенко предлагат уравнение включващо ролята на дифузията и по-пълно описващо процеса. B. Banerjee предлага емпирична формула за скоростта на реакция w (м3/ч на 1 м3 от катализатора) от различни показа-тели на процеса, отчитащи такива фактори като дифузия, стареене и отравяне на катализатора:
w = 8,26.106.e-4,38.103/T.P0,5P/250.0,2145.ѱ.(XH2O/0,37)0,5.4,5/d
, където 0,2145 – коефициент, отчитащ стареенето на катализатора и ди-фузията; ХН2О концентрация на водни пари в газа; ѱ – коефициент, отчитащ отравянето на катализатора със сяра (при изменено съдържание на серни съединения от 0,6 до 20 мг/м3 от сухия газ ѱ се изменя от 0,1 до 0,37); d – диаметър на частиците на катализатора.
Тази зависимост се използва за пресмятане на обема на зарежда-ния катализатор за висикотемпературна конверсия на въглероден оксид при известен срок на употреба на катализатора.
1.4 Метаниране
Газът, получен след конверсия на въглероден оксид и очистване от въглероден диоксид съдържа от 0,2 до 0,5% СО и до 1% СО2. Използване на този газ за различни процеси в нефтопреработката и нефтохимията е нерационално, а понякога и невъзможно. С цел очистване на газа от при-меси на въглеродни оксиди се използва реакциата на метаниране:
СО + 3Н2 →СН4 + Н2О + 206 kJ
CO2 + 4H2 →CH4 + 2H2O + 165 kJ
Принципните преимущества на метанирането се състоят в то-ва:
1. За провеждане на процеса не е необходимо на вход да се добавят допълнителни вещества; в газа подлагащ се на очистване се съдържа значително количество водород, което спомага за пъл-ното протичане на реакцията на метаниране.
2. В газа, който се подлага на очистване се съдържа СН4 и Н2O, следователно в процеса на метаниране не се вкарват вещест-ва, несъдържащи се в него до провеждането на този стадий.
3. Метанирането позволява едновременно да се очисти газа от примеси на кислород по реакцията:
Н2O + 0.5O2 →H2O + 242 kJ
Последното обстоятелство, което е съществено в този случай е ако охлаждането на газа на предходния стадий се осъществява с пара или впръскване на вода.
Реакцията на метаниране на СО е реакция обратна на ПК на метана и ТД й характеристики са дадени на стр. 12, а стойността на константите на равновесие са дадени в таблица 14:
Табл.14 Константи на
равновесие (К2) на реакцията СО + Н2О
![]() СО2 + Н2
СО2 + Н2
|
Температура, 0С |
К2 |
Температура, 0C |
К2 |
|
200 250 300 350 400 450 500 520 540 560 580 600 620 640 660 680 700 710 720 730 740 750 760 |
2,279.102 8,651.10 3,922.10 2,034.10 1,170.10 7,311 4,878 4,215 3,670 3,220 2,843 2,527 2,259 2,031 1,835 1,666 1,519 1,453 1,391 1,333 1,279 1,228 1,180 |
770 780 790 800 810 820 830 840 850 860 870 880 890 900 910 920 930 940 950 960 970 980 990 |
1,135 1,092 1,053 1,015 9,793.10-1 9,457.10-1 9,139.10-1 8,837.10-1 8,552.10-1 8,828.10-1 8,025.10-1 7,781.10-1 7.549.10-1 7,328.10-1 7,118.10-1 6,918.10-1 6,728.10-1 6,546.10-1 6,372.10-1 6,206.10-1 6,047.10-1 5,896.10-1 5,570.10-1 |
Константата на равновесие в реакцията на метаниране на СО2 се определя от уравнението:
Кр = рСН4.(рН2О)2
рСО2.(рН2)4
В температурния интервал 200 – 900 0С нейните стойности са след-ните:
Температура, 0С Кр Температура, 0С Кр
200 9,509.108 600 7,868.10-1
250 1,377.107 650 1,930.10-1
300 3,998.105 700 5,424.10-2
350 1,980.104 750 1,714.10-2
400 1,491.103 800 5,995.10-3
450 1,570.102 850 2,292.10-3
500 2,171.10 900 9,478.10-4
550 3,761
Равновесни концентрации на въглеродните оксиди могът да бъдат пресметнати, вземайки предвид уравнението за равновесните константи на реакциите и материалния баланс на процеса.
В условията на метаниране на газове получени в процеса на ПК, из-менението на концентрацията на водорода може да се пренебрегне. В то-зи случай пресмятането на равновесните концентрации на въглеродните оксиди значително се опростява и техните стойности могат да бъдат опре-делени с помощта на уравненията:
рСО = (рСО)2 рСО2 = 4.(рСО2)3
Кр.(рн2)3 Кр.(рН2)4
,където рСО, рСО2 и рН2 са парциалните налягания на компонентите на газа подлаган на метаниране.
Зависимостта на парциалните налягания на оксида и диоксида на въглерода в условията на ТД равновесие от температурата при съдържа-нието им в изходния газ в количество 1% е показана на фигура 9. При голям излишък на водород реакцията на метаниране при температури до 300 0С са практически необратими.
Повишаване на налягането е ТД благоприятно за протичане на реак-цията. Метанирането се провежда обикновено при 280 – 350 0С, налягане-то на процеса се определя от налягането на стадия на очистване от СО2, но ако полученият водород в последствие се компримира е възможно да се използва схема, предвиждаща метаниране при по-високо налягане. Обемната скорост зависи от налягането на процеса и използвания катали-затор и се колебае в границите от 1000 – 1500 ч-1, при атмосферно налягане до 6000 – 8000 ч-1 при 2Mpa.
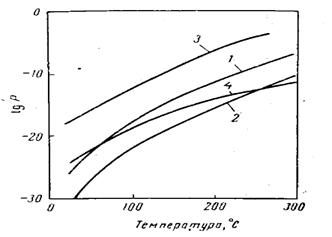

Съществен фактор при метанирането е отвеждането на отделящата се реакционнна топлина. Тук повишението на температурата на газа е 74 0С на 1% СО и СО2 встъпващи в реакция. Процесът е едностепенен и се провежда в адиабатен реактор. При тази концентрация оксидите на въгле-рода в газа, постъпващ за метаниране се ограничава от горната граница на работната температура на катализатора. Практически общото съдър-жание на въглеродни оксиди в изходния газ не превишава 1%. Отделянето на топлина се съпровожда също с реакция на възстановяване на катали-затора, обаче опасността от прегряване практически отсъства.
Хидрирането на въглеродните оксиди се осъществява с висока ско-рост върху катализатори изготвени на основа на метали от VІІІ група, но при производството на водород метанирането се извършва на никелов катализатор. В състава на катализатора влизат също носител (различни форми на алуминиеви и силициеви оксиди) и промотиращи добавки (нап-ример МgO, Cr2O3). В повечето случаи се използва катализатор във вид на таблетки.
Кинетическите закономерности на реакцията на метаниране на СО са изучени по-обстойно от метанирането на СО2. Предложено е уравнение за скороста на метаниране на СО в отцъствие на СО2.
w = k. (pH2)1/2.(pCO)3/2
(pCH4)
,където k – скоростна константа.
По други данни реакцията е от първи порядък по въглеродния оксид. Забелязано е, че при провеждане на реакция на метаниране скоростта на хидриране на СО2 е по-ниска отколкото при отсъствие на СО. Затова обик-новено в газа след метаниране остатъчното съдържание на СО2 е по-високо от съдържанието на СО. В реални условия провеждането на про-цеса, скоростта на тези реакции се забавя от дифузията и нейната зависи-мост от общото налягане на процеса се изразява с формулата:
w = k.P0,3…0,5
Табл. 15 Характеристика на катализаторите за метаниране
| Състав на катализатора |
Относи-телна плътност, кг/м3 |
Относи-телна повърх-ност, м2/гр |
Пори-стост, % |
Граница на издръж-ливост при натиск, N | |
| Съдър-жание на Ni,% | оксиди | ||||
|
50 50 40 18 50 50 50 40 – 50 |
Cr2O3 Al2O3 MgO Al2O3 +MgO SiO2 Al2O3 + SiO2 Cr2O3 + Al2O3 CaO + SiO2 + Al2O3 |
1200 1100 1000 1200 1000 1300 1200 900 |
145 140 140 15 150 130 140 100 |
50 55 52 53 50 45 50 57 |
44 98 284 147 196 167 88 196 |
След нискотемпературната конверсия на въглеродния оксид, газът постъпващ за метаниране не съдържа вредни за катализатора примиси. Действието на различните абсорбенти върху катализатора за метаниране е следното:
Адсорбент Действие
Воден разтвор на калиев Блокира порите на катали-
карбонат затора за метаниране при
изпарение на разтвора
Воден разтвор на калиев Същото действие, но ди-
карбонат + 3% диетанол етаноламина е безвреден
амин
Сулфолан, вода, диизо- Сулфоланът се разлага и
Пропаноламин предизвиква сярно отравяне
Моно- и диетаноламина Няма отровно действие
във воден разтвор
Метанол Същото
Друга причина за дезактивация на катализатора може да бъде него-вото прегряване от попадане на големи количества въглеродни оксиди вследствие от нарушаване на работния режим на стадия конверсия на оксидите на въглерода и промивка на конвертирания газ от СО2. Ако част от газа постъпващ за метаниране байпасира стадия на конверсия на СО е възможно отравяне на катализатора за метаниране със серни съединения. То е аналогично на отравянето на катализатора от частична-та конверсия.
2. Газификация на въглеводороди и нефтени остатъци
Процесът на газификация (частично окисление с кислород) на газо-образни и течни горива се осъществява във факел при температура 1300 – 1600 0С и налягане 3,0 – 10,2 MРa в стоманени реактори, футировани с огнеопорни материали. Газообразни и течни горива, кислород и пара се подават в реактора през горелка, където става разпрашаване на течното гориво на малки капчици и смесването им с окислителя. Капките гориво се изпаряват в атмосферата на горещия газ и взаимодействат с кислорода, образувайки факел. При частично окисление на пари и газове се изключва само стадия на разпрашаване и изпарение.
Процесът на газификация се осъществява при недостиг на кислород с образуване на горещ газ. В качество на окислител служи кислорода. Участието на другия окислител водните пари – в процеса на газифика-ция на въглеводородите е малко. Сярата се превръща в сероводород в повече от 90%. Около 0,5 – 3% от въглеродното гориво се превръща в сажди.
Полученият газ на 90 – 95% се състои от СО и Н2. В него се съдържа също въглероден диоксид, метан, азот, сероводород, органични съедине-ния на сярата, а също и непрореагирали водни пари. Необходимата дъл-бочина на превръщане без използване на катализатор се достига за сметка на провеждането на процеса при висока температура. Процесът се води при автотермични условия; топлината се получава за сметка на екзо-термичните реакции на газификация с образуване на СО и СО2.
Състава и добива на газа в процеса на паро-кислородна газифика-ция на алифатните въглеводороди се определя от условията на равнове-сие на същите реакции на парова конверсия на метана и конверсия на СО, които определят състава и добива на газ от ПК. Разликата е в това, че в реактора заедно с пара се подава и кислород, в който, макар и в неголеми количества се съдържа и азот. Уравненията на материалния баланс са дадени в таблица 16.
Газифията на алифатни въглеводороди с паро-кислородно подаване в производство на водород се използва рядко, тъй като е икономически неефективно в сравнение с паро-каталитичната конверсия. Практически интерес за производство на водород представлява газификацията на неф-тените остатъци – мазут, гудрон и др.
В състава на нефтените остатъци влизат не само високомолекулни парафинови въглеводороди, но и ароматни, хетероциклени и други съеди-нения. За технологични пресмятания на процеса на газификация е доста-тъчно да се знае елементния състав на суровината. Технологичните прес-мятания се улесняват и от това, че процеса се води при високотемперату-рни условия, когато съдържанието на метан в газа според условията на ТД равновесие е ниско и се определя основно от емпирични данни и лежи в границите 0,3 – 0,5% от газа.
При въвеждане на 0,5 м3 водна пара и 1 м3 кислород и достигане на равновесие не става отлагане на въглерод, но в действителност при про-цеса на газификация се отлага въглерод във вид на сажди. Затова в тех-нологичните изчисления се взема предвид саждообразуването в размер на 2 3%. За опростяване на технологичните изчисления се пренебрегва уравнието за равновесие на ПК на метана, а се отчита само равновесната реакция на конверсия на СО с водна пара.
Съдържанието на сяра в нефтените остатъци може да достигне 6 – 7%. ТД изчисления за преобразуването на сяра в условията на паро-кислородна газификация на нефтените остатъци са показали, че 90% от сярата се превръща в сероводород, около 7% - в карбамил-сулфид и 2% - във СS и неголямо количество – около 1% става на сажди.
Кислородът, съдържащ се в нефтените остатъци в количества не повече от 0,5 – 0,7 %, в процеса на газификация се превръща в кислород-съдържащи компоненти – Н2О, СО2 и СО. Не е нужно да се отчита вли-яние на кислорода в техническите изчисления, тъй като неговото присъст-вие практически не влияе на разхода на техническия кислород, нито на добива на газовите компоненти. Същото се отнася и за азота в суровината, съдържанието на който може да достигне до 1%. Азотът от суровината преминава основно в газа, но при газификация се образуват също в малки количества амоняк, азотни оксиди и цианиди. Трудно е да се определи за-висимостта на добива на тези съединения от съдържанието на азота в суровината. Технологичните изчисления, определящи разхода на кисло-род за газификация, добива и състава на газа за 1 кг суровина, се свежда до следното. Да обозначим елементния състав на 1 кг от суровината:
Въглерод............................. С Азот + кислород............ N
Водород.............................. Н Влага.............................. W
Сяра.................................... S Зола................................ А
Разходът на пара – „а” кг/кг от суровината, концентрацията на кисло-род в техническия кислород СО2, м3/м3, а концентрацията на азот, аргон и други благородни газове в техническия кислород тогава е 1 - СО2. Обема на кислорода в м3, изразходван за изгаряне на водорода δ, а степента на ПК на СО с образуване на Н2 и СО2 β.
За отределяне на въглерода, участващ в процеса на газификация с О2 и Н2О с образуване на СО + СО2, трябва от въглеродното гориво за извадим въглерода, превръщащ се в сажди (0,02 кг), и въглерод, израз-ходван за получаване на метан. Приемайки съдържанието на метана в га-за 0,5% и добива на газ – 3 м3 за 1 кг суровина, получаваме разхода на въглерод за образуване на метан равно на 0,008 кг. Количеството на гази-фицирания въглерод С` е равно на:
С` = С – (0,02 + 0,008)
Количеството на водорода в суровината, участващ в процеса на га-зификация се определя по уравнението:
Н` = Н – 0,00134 – 0,056.S
,където 0,00134 и 0,056.S е количеството водород, превръщащо се в СН4 и Н2S съответно.
Добива на отделните газови компоненти се определя по уравнения, приведени в таблица 16
Табл. 16 Материален баланс за пресмятане на паро-кислородната газификация на нефтените остатъци
| Компоненти |
Обем, м3 за 1 кг суровина |
|
| Изходни компоненти | Конвертиран газ | |
|
Н2О О2 N2 CO CO2 H2 CH4 H2S COS + CS2 |
1,243.(a + W) 0,935.C` + δ (0,935.C` + δ)(1 - СО2)/ СО2 - - - - - - |
1,243.(a+W)–1,867.C`.β+2.δ - (0,935.C`+δ).(1-СО2)/СО2+0,8N 1,867.C`(1 – β) 1,867.C`.β 11,2.H` + 1,867. C`.β - 2δ 0,015 0,63.S 100.S |
След заместване на обема на компонентите Н2, СО2, СО, Н2О от таблицата в уравнението на константата на равновесие за реакцията на конверсия на СО с водни пари ще получим:
K2 = CH2.CCO2
CCO.CH2O
K2 = β.(11,2.H` + 1,867.C`.β – 2.δ)
(1 β).[1,243.(a + W) – 1,867.C`.β + 2.δ]
Топлинният баланс на процеса на газификация се описва с урав-нението:
Qн + qт + qH2O + qO2 = 1,867.C`.(1 β).(12600 + t2.cCO) + (11,2.H` +
1,867.C`.β 2.δ).(11100 + t2.cH2) + [1.243.(a + W) + 2.δ]. t2cH2O +
1,867.C`.β. t2.cCO2 + qc + q
,където Qн е топлината от изгаряне на суровината, kJ/kg; qт е количество топлина, въвеждано със суровината в реактора, kJ/kg; qH2O е количество топлина, което се въвежда с парата, kJ/kg; qO2 е количеството топлина, което се въвежда с кислорода в реактора, kJ/kgl; t2 e температурата на из-ход от реактора, 0С; cCO, cH2, cH2O, cCO2 са средни топлинни капацитети на компонентите при постоянно налягане и температура на изход от реактора, kJ/(m3.0C); q – загуба на топлина в околната среда, kJ; qc – топлината на изгаряне и енталпията на саждите, метана, сероводорода, серните окиси, въглеродния сулфид и азота, пресметнати по уравнението:
qc = 1347 + 0,005.Qн + 18200.S
Решавайки съвместно уравнението на равновесие на реакцията на конверсия на СО с водни пари и топлинния баланс ще намерим β и δ.
Szargan, P. е изучавал процеса на паро-кислородна газификация на течно гориво в промишлен реактор. Реакционните зони в газгенератора, според неговите изследвания, са показани на фигура 10.
а б
Фиг. 10 а) Реакционни зони на факелния процес на газификация на течно гориво: 1 – зона на факела; 2 – зона на циркулация; 3 – зона на завършване на реакцията.
б) Изменение концентрацията на кислорода във факела при газификация на течни горива (прекъсната линия – екпериментални данни, непрекъсната линия – теоретични данни)
В горните зони течното гориво се разпръсква механично с форсун-ки и пневматично от паро-кислородната смес в горелката. Малките капки се изпаряват в атмосфера на горещ газ в зона 1 и 2. Потокът на окислителя засмуква изпареното гориво и газ от зона 2 в зона 1. В пламъка част от парите на горивото и газа изгарят, при което се достига температура по-висока от 2500 0С.
В противоположност на типичното направление на пламъка, този пламък е преобърнат, тъй като свободният кислород се намира отвътре, а горещият газ се засмуква отвън.
На фиг. 10 - б) е показано изменението на концентрацията на кисло-рода във факела за газификация на течни горива с отдалечаване от усти-ето. В пламъка, при концентрация на кислорода 2,2 м3 на 1 кг гориво, тем-пературата достига до 3000 0С. Заедно с продуктите от пълното горене СО2 и Н2О се образуват и продукти от дисоциация: СО, Н2, О2, а също и атомен кислород, водород и хидрооксиден радикал. Свободен и атомен кислород има не само на границите на пламъка, но и на разстояние, пре-вишаващо 1,6 пъти дължината на пламъка (концентрация на О2 > 1,2 м3/кг), а атомният водород и радикалът –ОН – на разстояние 2,2 (концентрация на О2 > 0,9 м3/кг). Дължината на пламъка се определя основно от сечение-то на изходната дюза на форсунката. Налягането в реактора не влияе на дължината на пламъка.
Обяснявайки химизма на процеса на газификация на течни горива и сравняването му с механизма на частично окисление на газообразно го-риво, авторите считат, че процесът протича в два стадия. Първо протича пълно изгаряне на въглеводородите, при което се изразходва всичкия кис-лород. Във втория стадий става конверсия на останалите въглеводороди с пара и въглероден диоксид. Предложен и механизъм на частично окисление на метана във факела с отчитане образуването на ацетилен в качеството му на междинен продукт. Съобразно с този механизъм проце-сът протича на три стадия: на първия става верижна реакция на окисле-ние на метана и се образуват предимно въглеводороди С2 и вода, на вто-рият стадии има окисление на ацитилена и водорода, а на третия – пълно окисление на всички междинни продукти до въглероден оксид и водород.
Л.Н. Алейнова и колектив експериментално определят основните стадии на непълно горене на метана. Авторите считат, че процесът се състои от три стадия. Първият се характеризира с пълно отреагиране на кислорода и натрупване на СО, водород, СО2, вода и малки количества ацетилен; на вторият стадий се натрупва ацетилен и спира натрупването на водни пари и СО2; на третият стадий става конверсия на въглеводоро-ди, разграждане на ацетилена до отделните елементи и газификация на саждите.
Отличителна особеност на газификацията на течни горива е отделя-не на сажди. Газификацията се води при условия, водещи до образуване на сажди, въпреки че при достигане на ТД равновесие, въглеродът в продуктите на реакцията трябва да отсъства. Получаването на сажди зависи от относителния разход на кислород и водна пара, температурата и налягането на процеса, качеството на разпръскане на горивото и смес-ването му с окислителя. Получаването на сажди намаля с увеличаване от-носителния разход на кислород и водна пара. Установено е, че при отно-сителен разход на кислород, равен на 0,6 м3 на 1 кг мазут, се отделя 7% (масови) сажди (изчислено по мазута), при относителен разход 0,8 м3/кг 0,5% (масови).
Образуването на дисперсен въглерод се обяснява с това, че заедно с процесите на непълно и пълно изгаряне при газификация протичат процеси на разцепване на въглеводородите. В съответствие с основните положения в теорията на П. А. Теснер, образуването на сажди се предшества от образуването на радикал, от който се образува зародиша на саждената частица. От съотношението кислород:гориво зависи не само добива на сажди, но и тяхната относителна повърхност.
Електронно-микроскопски изследвания на сажди, получени от паро-кислородна газификация на мазут, показали, че първичните частици имат сферична форма и диаметър 20 - 50ηm. Има и по-големи частици, които, както предполагат авторите, се образуват от механично съединяване на първичните. От това изследване се прави извод, че саждите в процеса на газификация на течни горива се образуват от паровата фаза, а не от теч-на при коксуване на капки от гориво, както се е предполагало по-рано.
Електронно-микроскопските изследвания показали също, че първич-ните частици изгарят отвън и с повишение на температурата стават силно пористи. Някои частици се окисляват до търкостенни черупки, защото окислението при тях протича първо във вътрешността на частицата.
Изборът на режим на газификация на течните и газообразни въгле-водороди се определя не само от условията на ТД равновесие, отколкото от техническите възможности за водене на процеса. Давлението се под-бира в зависимост от технико-икономическите разчети. Широко разпрост-ранение са получили инсталации за газификация при налягане 2,0 – 4,2 MРa; максималното налягане на процеса може да достигне 10 Mрa.
В зависимост от условията на ТД равновисие, ниска концентрация на метан в газа може да се достигне при температура по-висока от 1000 0С. Но скоростта на реакция при такава температура без катализатор е малка, а при това се наблюдава значително саждообразуване. Затова процесът се провежда при температура по-ниска от 1300 0С. С повишава-не на налягането трябва да се повиши и температурата на процеса, например по следния начин:
Налягане, МРа............................ 3 6 10
Температура, 0............................ 1300 1350 1400
Отношението пара:суровина се приема също от емпиричните данни от 0,3 до 0,5, при което колкото по-тежка е суровината, толкова по-висок е разхода на пара. Подгряването на изходните компоненти позволява малко да се намали разхода на кислород и да се подобрят условията на проце-са.
Температурата на подгряване на суровината се колебае в широки граници от 80 до 300 0С. Минималната температура е ограничена от вис-козността на суровината: суровината трябва да бъде подгрята минимум до 20 – 30 0С, като при тази температура визкозитета става достатъчен за по-даване и разпръскване във форсунките. В тях суровината се разпръсква добре при вискозитет 3 – 4 условни градуса. Максимално допустима тем-пература на нагряване на суровината се ограничава от условията на отде-ляне на кокс по стените на подгревателя на суровината. Суровината ряд-ко се подгрява в подгреватели с пряк огън, а с водна пара с ниско наляга-не и температура 110 – 120 0С.
Най-сложно се явява подгряването на кислорода във връзка с ръста на неговата реакционна активност по отношение на металите. Температу-рата на кислорода, подаван в реоктора, се избира в границите от 20 – 300 0С. Безопасното подгряване на кислород до 300 0С може да се обезпечи при използване на прегрята водна пара.
Разходът на кислород зависи също и от загубите на топлина в реак-ционната зона, а последните зависят от производителността и конструк-тивното оформление на газгенератора и е в границите 0,1 – 4% от топли-ната на изгаряне на изходната суровина. В съвремените високопроизводи-телни промишлени газгенератори загубата на топлина в реакционната зона не превишава 0,3 – 0,5%.
В таблица 17 са приведени показатели на процеса паро-кислородна газификация на природен газ, бензинови пари и тежко котелно гориво при налягане около 3 МРа, а в талица 18 – газификация на различни суровини про налягане 6 МРа по данни на фирма Shell. В таблица 19 е даден мате-риален баланс от газификацията на мазут.
Табл. 17 Показатели на процеса паро-кислородна газификация на въглеводороди при налягане 3 МРа
| Показатели | Приро-ден газ | Бен-зин | Тежко котелно гори-во |
|
Разход за 1 кг от суровината кислород (95%-ен)............... водна пара (с температура на насищане 246 0С)........... Температура на подгряване, 0С суровина.............................. кислород.............................. водна пара.......................... Добив на газ, м3 за 1 кг суровина Състав на газа, % СО2 ………………………….. СО …………………………… Н2 ……………………………. СН4 ………………………….. N2 + Ar ………………………. H2S + COS . |
0,98 0,05 236 236 246 3,73 2,8 34,5 61,0 0,3 1,4 - |
0,83 0,35 300 300 300 3,29 3,9 41,7 52,7 0,3 1,4 7.10-3 |
0,75 0,40 236 236 246 2,95 4,3 46,9 46,2 0,3 1,4 0,9 |
Табл. 18 Показатели за процеса паро-кислородна газификация на Различни суровини при налягане 6 МРа
| Показатели | Природен газ | Лек бензин от пър-вична дестилация | Мазут | Крекинг-остатък | Вакуум остатък | Остатък от деас-фалтизация | Мазут | Крекинг-остатък |
|
Температура, 0С, на вход в реактора суровина............... кислород............... пара....................... Разход на кислород за 1 кг суровина (95%) м3/кг....................... пара, кг.................. газ (сух), м3........... Състав на газа, % СО2........................ СО......................... Н2.......................... СН4....................... N2.......................... Ar.......................... H2S....................... COS..................... Производство на пара в котел-утилизатора, кг/кг............................ Налягане на насите- ната пара, МРа........ Добив на сажди, % (тегл.) |
Без циркулация на сажди |
С циркула- ция на саж- ди |
||||||
|
300 245 313 0,983 0,20 3,63 3,88 33,82 60,13 0,60 0,72 0,85 - - 3,54 10,5 0,02 |
250 245 313 0,879 0,40 3,21 5,21 41,29 51,45 0,60 0,58 0,86 0,01 - 3,06 10,5 3,0 |
245 245 313 0,750 0,45 2,94 4,91 46,19 46,02 0,60 0,65 0,80 0,80 0,03 2,72 10,5 3,0 |
245 245 313 0,763 0,45 2,98 4,86 46,89 45,81 0,60 0,63 0,81 0,38 0,02 2,81 10,5 3,0 |
245 245 313 0,745 0,50 2,90 5,55 46,54 45,03 0,60 0,68 0,81 0,76 0,03 2,80 10,5 3,0 |
300 245 313 0,771 0,55 2,80 6,09 46,99 43,26 0,60 0,62 0,80 1,57 0,07 2,80 10,5 3,0 |
245 245 313 0,776 0,45 3,01 5,10 46,61 45,42 0,60 0,65 0,81 0,78 0,03 2,85 10,5 3,0 |
245 245 313 0,786 0,47 3,06 5,15 47,01 45,41 0,60 0,63 0,81 0,37 0,02 2,90 10,5 3,0 |
|
Табл. 19 Материален баланс на паро-кислородната газификация на мазута
| Компоненти |
Коли- чество |
Тук се включват елементите | ||||
| С | Н | О | N | S | ||
| Приход, кг | ||||||
|
Мазут........................... Водна пара................. Кислород..................... |
100,0 46,0 100,9 |
87,3 - - |
9,8 5,1 - |
0,8 40,9 100,9 |
0,7 - - |
1,4 - - |
| Общо...... | 246,9 | 87,3 | 14,9 | 142,6 | 0,7 | 104 |
|
Н2................................. СО……………………… СО2 ……………………. Н2О ……………………. Н2S.............................. COS............................. N2................................. CH4.............................. Сажди.......................... |
12,0 178,0 26,9 24,1 1,4 0,1 0,7 0,4 3,3 |
- 76,4 73 - - 0,02 - 0,3 3,3 |
12,0 - - 2,7 0,1 - - 0,1 - |
- 101,6 19,6 21,4 - 0,02 - - - |
- - - - - - 0,7 - - |
- - - - 1,3 0,1 - - - |
| Общо....... | 246,9 | 87,3 | 14,9 | 142,6 | 0,7 | 1,4 |
3. Методи за отделяне на водорода от водородсъдържащи газове
Нефтозаводските газове представляват смес от въглеводороди и водород. Основните физични константи на водорода и на газообразните въглеводороди са дадени в таблица 20. Водородът се отделя от тези газо-ве чрез методите: дълбоко охлаждане, абсорбция, адсорбция, дифуция през мембрани с избирателна пропускливост. Криогенният метод е наме-рил промишлено приложение за отделяне на Н2 от водородсъдържащи газове. За получаване на водород с висока чистота се използва метода на адсорбция при променливо налягане (PSA – pressure swing adsorption, КЦА – короткоцикловая адсорбция), върху зеолити. Водород с висока чистота се получава в малки количества при дифузия на през мембрани от сплави на паладия, проницаеми спрямо водорода, но непроницаеми за други газ-ове и пари. Разработват се и полимерни мембрани притежаващи подобни свойства. Методът за абсорбция с въглеводороди и следваща ректифика-ция, особенно при понижена температура, също може да бъде използван за концентриране на водорода. Посочените процеси може да се използват не само за отделяне на Н2 от нефтозаводски газове, но и за отделянето му в различни процеси за производство на водород. Доколкото способите и принципите са аналогични, отделянето на водорода и неговата доочистка се разглежда във всеки раздел съвместно с отделянето на водорода от нефтозаводските газове.
Табл. 20 Физични константи на газове, влизащи в състава на нефтозаводските газове
| Газ |
Плътност при 0 0С и 0,1 МРа, кг/м3 |
Молекулна маса | Т. кип. при 0,1 МРа, К | Газова кон-станта R, J/(kg.K) |
Специфична топлина на кипене, kJ/kg |
Критични константи | |
|
Ткр, К |
Ркр, МРа |
||||||
|
Н2................. СН4............... С2Н6.............. С2Н4.............. С3Н8.............. С3Н6.............. n-C4H10 …….. n-C4H8 ……… n-C5H12 .. |
0,09 0,717 1,356 1,260 2,005 1,914 1,703 - - |
2,0159 16,04 30,07 28,05 44,09 42,08 58,12 56,104 72,146 |
20,38 111,67 184,53 169,37 231,1 225,45 272,65 266,66 309,21 |
4124,2 518,2 276,7 296,6 188,5 197,5 143,0 - - |
451,8 510,4 489,9 483,2 448,0 427,0 386,5 404,0 483,2 |
33,08 190,1 308,27 282,5 369,81 365,0 425,01 417,0 470,2 |
1,30 4,62 4,89 5,14 4,26 4,59 3,80 4,02 3,35 |
3.1 Криогенен метод за концентриране на водорода
Отделяне на водорода от негови смеси с газообразни въглеводоро-ди чрез метода на фракционна кондензация се провежда с охлаждане на газовата смес до температури, при които въглеводородите преминават в течно състояние, а водородът остава в газообразно. Разделянето се осно-вава на разлики в парциалните налягания на водорода и въглеводороди-те. Концентрирането на водорода представява по същество задача свър-зана с отделянето на водорода от метана, доколкото другите въглеводоро-ди кондензират при по-високи температури от метана. Равновесието на системата водород – метан се определя от режимните условия по темпе-ратура, необходими за получаване на водород с необходимата степен на чистота. На фигура 11 е показана зависимостта на концентрацията на по-лучавания водород от температурата при различни налягания. Конденза-цията на метана се води при 2 6 МРа. Както се вижда от фигурата, водо-род с концентрация 95% може да се получи при налягане 2 МРа и темпе-ратура минус 166 0С, а при 6 МРа при температура минус 158 0С. За по-лучаване на водород с по-восока концентрация е необходима по-ниска температура.
В областта на използваните налягания, разтворимостта на водорода в течен метан не е голяма, което се вижда от следните данни:
Температура, К Налягане, МРа Съдържание на Н2, % (мол)
в течната фаза в газовата фаза
90,3 4,84 2,4 96,8
7,50 3,6 97,5
103,1 3,85 2,2 94,7
7,60 4,0 96,4

3.2 Отделяне на водорода с помощта на полупроницаеми мембрани
Мембрани, проницаеми за водород и непроницаеми за други газове се използват в някои отрасли на промишлеността за получаване на свръх чист водород.
Механизмът на проникване на водород през паладиева преграда е следния. Водородът се адсорбира на повърхността на метала и се дисо-циира с образуване на атомен водород. Последният, отдавайки електрон на паладия, преминава в положително зареден протон. Протонът преми-нава през слоя на паладиевата атомна решетка и присъединявайки елек-трон образува отново неутрален атом на водорода. Атомите на водорода от се съединяват в молекули и десорбират от повърхността в газова фаза. Проникването на Н2 приз паладиевата мембрана не представлява типичния дифузионен процес.
Чистият паладий не издържа на налягане, а се пука и разрушава в среда от водород, затова са направени много изследвания по подбора на сплави на паладия с други метали. Днес съществуват сплави с по-висока якост, устойчиви в среда на водород и при наличие на такива примеси като СО, СО2, Н2О и въглеводороди С1 – С5, поради което проницаемостта на водород пред сплави на палодий е по-висока, отколкото през чист пала-дий. Такива сплави, обаче, са неработоспособни при наличие на серни съединения в газа. Добра проницаемост и висока устойчивост показали сплавта на паладий със сребро и никел (85% Pd, 10% Ag, 5% Ni), сплав на паладия със сребро, иридий и платина (66% Pd, 31% Ag, 3% Ir, 0,2% Pt). Има предложения с цел да се намали стойността на сплавта да се замени среброто с мед.
Колкото по-тънка е паладиевата преграда, толкова повече водород може да премине през нея и в същото време по-малко са разходите за оборудване с този скъп метал. Изготвят се дискове от пориста керамика и на тях се нанасят глазури от 91% Pd или сплав на паладия със сребро с последващо изпичане. В електронните приспособления са разработени много способи за нанасяне на свръхтънки филми: катодно напрашаване, електролиза, пиролиз на металоорганични съединения и др.
Една от най-характерните формули за пресмятане на проницаемост-та на водорода е:
w = [K1.(10.p1)n – K2.(10.p2)m].e-E/RT.1/δ
, където w – пронецаемост, м3/(м2.ч); δ – дебелина на мембраната, мм; р1 и р2 – парциални налягания на Н2 на и след мембраната, МРа (р1 > р2); К1, К2 коефициенти; Е – енергия на активация, kJ/mol; R – газова константа, kJ/(mol.K); n, m – степенни показатели.
За сплав от 85% Pd, 10% Ag и 5,5% Ni се препоръчва да се приеме Е = 13400 kJ/mol.
При р1 > 1,2: n = 0,78; m = 0,85; K1 = 0,87; K2 = 0,96
При p2 > 1,2: n = 0,63; m = 0,68; K1 = 1,26; K2 = 1,20.
Освен мембраните от паладиеви сплави, които се използват в интер-вала 200 – 700 0С, са разработени и полимерни мембрани, пропускащи во-дород и задържащи другите газове. Използват се например снопове от ку-хи полиетерни влакна с вършен диаметър 36 мкм при вътрешен диаметър 18 мкм за отделяне на Н2 от водородсъдържащи газове в нефтопреработ-вателните заводи. Сноп с диаметър 300 мкм има около 32 млн. Такива влакна. Газът навлиза в каналите на влакната, водородът преминава през стените и се извежда от пространството между влакната.
3.3 Абсорбционна очистка на газове в процеса на производство на водород
3.3.1 Изисквания към системата за очистка на газове
Абосорбционната очистка на газове се използва в производството на водород чрез метода на паро-каталитична конверсия и паро-кислородна газификация на въглеводороди. При получаване на водород чрез метода на паро-каталитична конверсия на въглеводороди, газът след конверсия на СО се подлага на очистване от СО2. В газа след конверсия, както се вижда от таблица 21, се съдържа до 23% СО2 и практически отсъстват серни компоненти. Общото налягане в системата за конверсия на СО е 1,6 – 2,0 МРа. По такъв начин парциалното налягане на СО2 на вход в абсор-бера е равно на 0,28 – 0,46 МРа. При производство на водород под ниско налягане парциалното налягане на въглеродния диоксид пред очистката е около 0,02 МРа. Предложени са схеми, предвиждащи компресиране на газа след конверсия до това налягане, при което ще бъде използван.
Табл. 21 Съдържание на СО2 и Н2S в сухите газове до и след очистка в инсталациите за производство на водород
|
Съдържание до очистка |
Съдържание след очист-ка | |||
|
СО2, % |
H2S, % |
CO2, % |
H2S, мг/м3 |
|
|
Паро-каталитична конверсия на въглеводороди Газ след конверия на СО.................... 16 - 23 - 1,5 - 0,1 - Паро-кислородна газификация на нефтени остатъци Газ след газификация*........................ 4 – 6 0,1 – 1,0 неогнани- до чено 30 - 50 Газ след конверсия на СО*................ 33 – 34 - 0,2 – 0,8 - Газ след конверсия на СО*................ 33 – 34 0,1 – 1,0 0,2 – 0,8 1 - 2 |
||||
* схема с котел-утилизатор
* схема с впръскване на вода
В производството на водород по метода на паро-кислородна газифи-кация на нефтените остатъци трябва да се проведе абсорбционна очистка на газовете не само от СО2, но и от H2S и други серни съединения. В схе-ма с котел-утилизатор след газогенератора охлаждения и очистения от сажди газ се полага на сероводородна очистка по абсорбционния метод, а след конверсията на СА се провежда очистване от СО2. Газът постъпващ за сероочистка съдържа 0,1 – 1% сероводород и 20 – 600 мг/м3 органични съединения на сярата, главно карбамил сурфид. В газа се съдържа също 4 – 6% СО2, отделянето на който на стадия на очистване от сероводород технологично не е необходимо да се провежда, но въпреки това става пог-лъщане, в една или друга степен. След конверсията на СО, газът съдържа до 34% СО2.
Паро-кислородната газификация на нефтените остатъци с охлажда-не на газа в котел-утилизатор при налягане 3 – 6 МРа.Парциалното наля-гане на H2S преди очистване може да варира от 0,002 до 0,07 МРа, а пар-циалното налятане на СО2 след конверсия на СО е от 0,83 до 2,4 МРа.
Производство на водород по метода на паро-кислородна газифика-ция на нефтените остатъци се осъществява и без котел-утилизатор. В този случай газа се охлажда за сметка на впръскване на вода в количество, осигуряващо също и очистване от сажди. Наситеният на водни пари газ, съдържащ серни съединения, постъпва за среднотемпературна конверсия на въгледороден оксид. След конверсията на СО, газа се очиства от СО2 и сероводород. Процесът се води при налягане 11 – 17 МРа. Газът, постъп-ващ за очисване е с парциално налягане на СО2 от 3,6 до 5,8 МРа и на сероводород от 0,011 до 0,17 МРа.
Газовете, подавани за получаване на сяра, трябва да съдържат не по-малко от 10 – 15% Н2S, а останалото СО2. Примесите Н2S в отделе-ния от газа въглероден диоксид трябва да бъдат минимални, за да е въз-можно изхвърляне на СО2 в атмосферата, без тя да се нарушават сани-рните норми. Задачата е сложна, тъй като в газа след газификация се съдържа 6-20 пъти повече СО2, отколкото Н2S, а в газа след конверсия на СО – 30 60 пъти повече отколкото Н2S.
Препоръчва се процеса да се води за сметка на топлинната енергия, съдържаща се във влажния конвертиран газ. Използването на нископотен-циална топлина (от нереагиралите водни пари) за процеса може същест-вено да снижи разходите.
Съдържанието на въглероден диоксид в сухия очистен газ не трябва да превишава 0,1 – 0,2%. Използването, обаче, на един от най-разпрост-ранените методи на очистване – с горещ разтвор на К2СО3 (поташ) без активни добавки, не позволява да се достигне такава дълбочина на очистка и в сухия газ остава до 0,8% СО2. Повишаване на неговото съдържание води до допълнителен разход на водород в процеса на метаниране и увеличено съдържание на метан в получения водород. Преимуществата на очистването с горещ разтвор на поташ са толкова го-леми, че без да се има предвид на преразхода на водород, този метод се използва широко.
В схемите за производство на водород, които не предвиждат мета-ниране на СО или обезпечаващи селективно метаниране на на въглерод-ните оксиди се допуска по-груба очистка – до 1,5% СО2. Тя може да бъде при производство на водород под налягане, ако водородът е предназна-чен за хидроочистка на сулфидни катализатори; тук СО2 не взаимодейства с водорода, а само понижава неговото парциално налягане. Невстъпвайки в реакция, СО2 се разтваря в хидрогенизата.
В схемите на паро-кислородна газификация на нефтени остатъци с впръскване на вода след газогенератора, газът след конверсия на СО трябва да е практически напълно освободен от серни съединения. Това се налага, защото газа ще бъде подаден на никелов катализатор за метани-не (чувствителен към отравяне със серни съединения) или в системата за медно-амонячна очистка от СО.
В процесите на производство на водород се случва да се очиства газ с различни кисели компоненти при много различни пациални налягания на тези компоненти. В производството на водород се използват абсорбцион-ни способи за очистка химични, физични и комбинирани поглътители.
3.3.2 Основни принципи на абсорбционната очистка
Очистка на газа от СО2 и H2S се извършва с течни поглътители в абсорбер, а после тези компоненти се отделят от газа в десорбер. Проц-сът на абсорбционна очистка е цикличен. Поглъщането се основава на хи-мически взаимодействия със слаби базистни свойства и образуване с тях на неустойчиви съединения. Другите компоненти на газовата смес, непри-тежаващи киселинни свойства, не се поглъщат от течността. На стадия на регенерация, в резултат на повишената температура на поглътителя и снижаване на парциалното налягане на погълнатите компоненти, химичес-ките връзки се разрушават.
Очистване на газове от въглероден диоксид и сероводород, с поглъ-тител разтварящ газа, в цикличен процес се основава на изменяне на на-лягането в абсорбера и регенератора, за което не се изисква разходване на топлина и охлаждаща вода. Отначало в качество на абсорбент се е използвала вода, а по-късно били подбрани органични поглътители. Веще-ства, химически свързващи СО2 и Н2S, разтварят или се смесват с химиче-ски неутрални течности, например вода и действат също като поглътите-ли. Затова всеки химически поглътител е в една или друга степен абсор-бент. Изборът на поглътител се определя от налягането на абсорбцията. Физически поглътители се използват само при високи налягания.
Химични поглътители позволяват да се получи по-висока степен на очистка, отколкото физическите, но са необходими енергийни разходи за разрушаване на образуващите се съединения. По-голяма дълбочина с по-малки разходи на топлина може да се получи, комбинирайки два типа поглътители: леко регенериращи се за груба предварителна очистка и по-трудно регенериращи се за по-дълбока очистка.
Константата на фазово равновесие или коефициента на разпределе-ние на газа между газова и течна фаза се определя по формула:
KCO2 = pCO2:x KH2S = pH2S:y
, където pCO2, pH2S – парциални налягания на СО2 и Н2S над разтвора, МРа; x, y – количеството СО2 и Н2S, разтворено в течността, м3/м3.
Уравнението на фазово равновесие с отчитане зависимостта на коефициента на температурно разпределение лежи в основата на циклич-ната очистка на газове от СО2 с течни поглътители. Коефициента на раз-пределение и неговото изменение в зависимост от температурата е основ-на характеристика на поглътителя.
Кривата на разпределение, тоест зависимостта на количеството на газа, погълнато от течността, от парциалното налягане на газа на течност-та pCO2, pH2S се опледеля за всеки поглътител експериментално. Общият характер на кривите на разпределение е показан на фигура 12, от която е видно, че поглътителя имащ крива на разпределение 1 е по-малко ефек-тивен от този с крива на разпределение 2, тъй като неговата поглъща-емост при едно и също парциално налягане. За достигане на тази степен на очистка регенерацията на поглътител 1 трябва да е по-дълбока (х``1<x``2). Подгряването на поглътителя в регенератора до t``1 позволява да се достигне необходимата дълбочина на регенерация х``1 при по-висо-ко парциално налягане на СО2 в регенератора и да се създадат условия, при които парциалното налягане на СО2 по-високо от равновесното, тоест ррСО2>р``СО2.
Принципната схема на абсорбционна очистка на газа от СО2 и Н2S с нагряване на поглътителя в процеса на регенерация е показана на фигура 13. Газът се въвежда в абсорбер 1 отдолу и след очистване излиза отгоре. Регенерираният поглътител с температура t1, близка до температурата на върха на абсорбера, се подава отгоре. Поглътителят, наситен на въглеро-ден диоксид, се извежда от абсорбера и преминава през топлообменник 3, където се подгрява за сметка на топлината на регенерирания поглътител. Ако процесът протича при по-високо налягане, отколкото налягането в регенератор 6, то понижаване налягане на поглътителя се извършва, като го прекарват през турбина 5; при това от поглътителя се отделя част от СО2.
По-нататък поглътителят с разтворен в него СО2 постъпва на върха на регенератора 6. В долната част на регенератора поглътителят се нагря-ва с помощта на ребойлер 7 и тук завършва регенерацията. Регенерира-ният абсорбент се прекарва посредством помпа през топлообменник 4 и хладник 3 и хладилник 2 в абсорбера.


© 2009 База Рефератов