
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
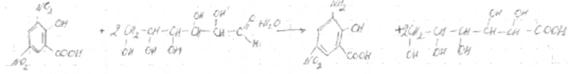
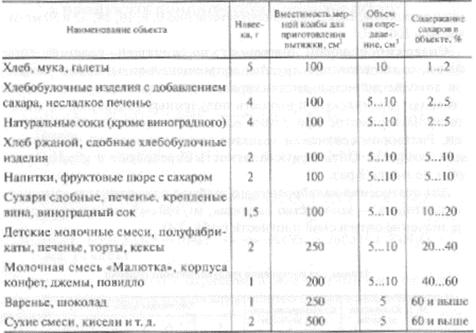
Контрольная работа: Химические методы определения сахаров
Контрольная работа: Химические методы определения сахаров
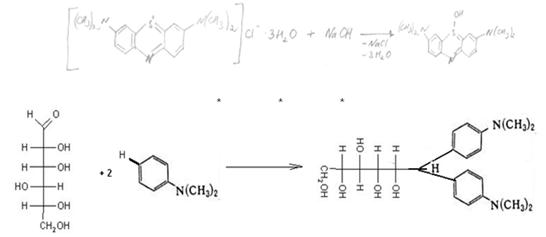
Общее представление о веществах, обьединяемых под названием «сахара»
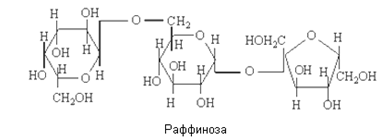
Пищевые продукты содержат главным образом дисахариды (сахароза, мальтоза, лактоза), моносахара (глюкоза, галактоза, фруктоза); из трисахаридов — в основном раффинозу. Для большинства продуктов нормируется суммарное содержание сахаров (общий сахар), а для некоторых веществ (карамель, патока и др.), кроме того, содержание редуцирующих сахаров, т. е. сахаров, способных легко окисляться. Все перечисленные выше сахара (за исключением сахарозы) обладают редуцирующей способностью. Общий сахар — это суммарное содержание сахарозы и восстанавливающих низкомолекулярных сахаров, выраженное в процентах сахарозы. В последние годы термин «сахара» применяют только по отношению к моносахаридам.
Молекулярная интерпретация химических — аналитически значимых — свойств сахаров
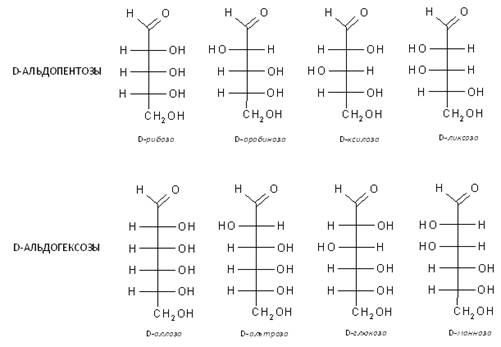
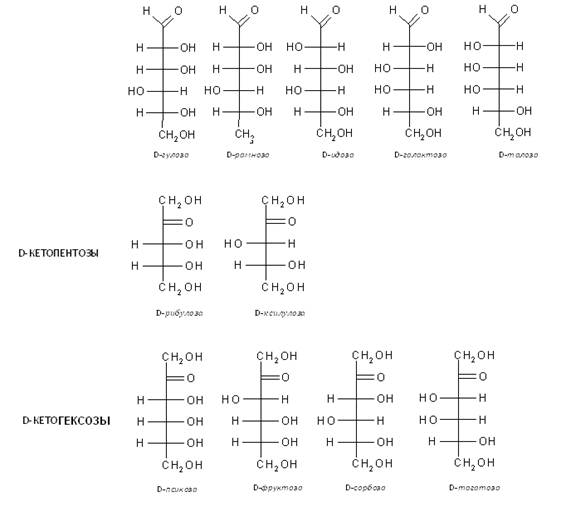
В природе моносахариды (монозы) распространены и имеют наибольшее значение пентозы общей формулы С5Н10О5 и гексозы общей формулы С6Н12О6, не могут гидролизоваться и превращаться в более простые углеводы. По химическому строению моносахариды представляют собой многоатомные спирты, имеющие альдегидную группу (альдегидоспирты, оксиальдегиды, альдозы — когда на конце углеродной цепи присутствует карбонильная группа) или кетонную группу (кетоспирты, оксикетоны, кетозы — когда карбонильная группа расположена в ином другом положении). Отсюда, глюкоза и фруктоза являются функциональными изомерами. Функциональные изомеры различаются и расположением гидроксильных групп, как это видно из сравнения структур глюкозы и галактозы. Такие пространственные изомеры называются диастереомерами. Они различаются физическими и химическими свойствами.
Для
моносахаридов характерна оптическая изомерия (энантиомерия) (Э. Фишер). В их
молекулах содержатся асимметрические (хиральные) атомы углерода (С*),
находящиеся в sp3-гибридизации
и связанные с четырьмя различными атомами или их группами. Энантиомеры имеют
идентичные физические и химические свойства. Число оптических стереоизомеров
связано с числом асимметрических атомов углерода формулой N
= 2n. ![]()
![]() В общем случае молекула с "n" хиральными
центрами имеет 2n стереоизомеров,
которые представляют собой пары зеркальных антиподов.
В общем случае молекула с "n" хиральными
центрами имеет 2n стереоизомеров,
которые представляют собой пары зеркальных антиподов.
В качестве обозначения противоположных конфигураций редуцирующих сахаров с одинаковыми названиями применяются буквы D и L, которые указывают принадлежность к двум рядам. Последний асимметрический атом углерода определяет принадлежность к стереохимическому ряду. Гидроксильная группа справа – D ряд; слева – L – ряд. В природе одни моносахариды встречаются больше в Д-конфигурации, чем в L, другие наоборот (L-арабиноза часто встречается в растениях, тогда как D-арабиноза обнаружена только в некоторых видах бактерий).
Поскольку молекулы сахаров построены несимметрично, то для них не может быть оптически неактивных мезоформ. Экспериментальный факт, фиксируемый поляриметром, вращения поляризованного света вправо обозначается знаком (+); вращение влево — знаком (-). Символы знаков D(+) – и L(-) могут очень часто не совпадать.
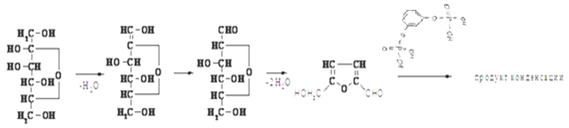
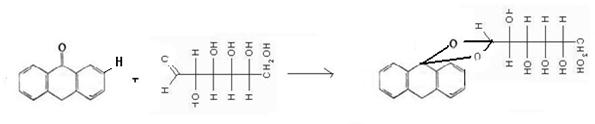
Изображенные линейными формулами альдозы во многом не отражают физические и химические свойства моносахаридов. Такие структуры возможны в растворах, и то в незначительных количествах (тысячные доли процента). Так в УФ - и ИК-спектрах моносахаридов отсутствуют полосы поглощения, характеризующие карбонильную группу. В действительности, моносахариды с 5 и более атомами углерода обычно встречаются в водном растворе в циклических формах, в которых карбонильная группа образует ковалентную связь с атомом кислорода гидроксильных групп. А.А. Колли (русский химик-органик), предложил для всех моносахаридов циклические структуры.
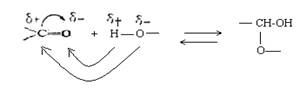
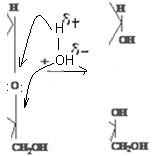
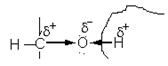
В карбонильной группе связь между атомами углерода и кислорода осуществляется двумя парами электронов. Электронное облако связи смещено к кислороду как более электроотрицательному атому, в результате чего он приобретает частичный отрицательный .заряд (δ-). В то же время карбонильный углерод в результате оттягивания от него электронов приобретает частичный положительный заряд (δ +):

Это обусловливает большую реакционную способность органических соединений, содержащих карбонильную группу: с одной стороны, атом углерода приобретает электрофильные свойства и способен активно взаимодействовать с нуклеофильными реагентами, а атом кислорода приобретает нуклеофильные свойства и способен присоединять электрофильные реагенты и замещаться ими; с другой стороны, на атоме углерода связи С=О концевой карбонильной группы положительный заряд сильнее притягивает электрон от атома водорода альдегидной группы, последний становится более подвижным и легко вступает в какие-либо реакции, например, окисляется (редуцируется) при рН > 7.
Отсюда, появление пятого гидроксила, названного гликозидным, является следствием внутримолекулярной реакции между карбонильной группой и одним из гидроксилов сахара. Известно, что взаимодействие альдегидов (кетонов) со спиртами приводит к образованию полуацеталей (полукеталей):
На связь между устойчивостью цикла и его строением обратил внимание немецкий химик А. Байер. В своей теории он исходил из предположения, что все циклы являются плоскими, а за меру устойчивости цикла принял любое отклонение валентных углов от "нормального" угла 109° 28':
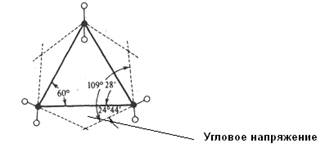
Такое отклонение создает в молекуле напряжение, которое, в свою очередь, понижает ее устойчивость. Расчет дал отклонение валентного угла от нормального для четырехчленного цикла +9°44', пятичленного 0°44', шестичленного -5° 16'.
Также позже было установлено, что пяти- и шестичленные циклы не находятся в одной плоскости, а принимают в пространстве определенную форму (конформацию: см. ниже «кресло»: она устойчивее), что приводит к уменьшению углового напряжения, поэтому и к повышению устойчивости цикла.
Поэтому при образовании циклических структур сахаров-альдоз гидроксильная группа у предпоследнего (или пред-предпоследнего) реагирует с альдегидной группой, образуя пиранозный (или фуранозный) цикл, содержащий полуацетальную связь:
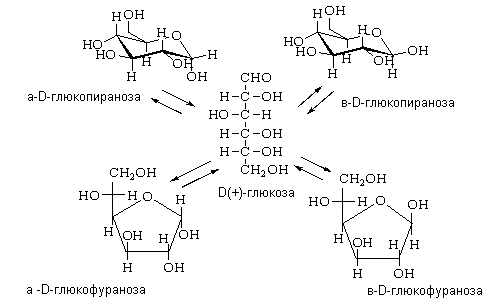
Сахара-кетозы также встречаются в форме α- и β-аномерных форм. В этих соединениях гидроксильная группа у предпоследнего (или последнего) реагирует с кетогруппой, образуя фуранозный (или пиранозный) цикл, содержащий полукетальную связь:
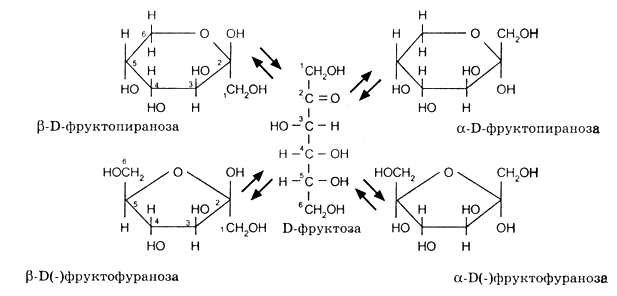
При образовании циклического полуацеталя сахаров появляется новый асимметрический атом углерода (аномерный центр (С-1)). Для указания его конфигурации используют обозначения a и b. α- и β-формы моноз превращаются друг в друга в водном растворе, этот процесс получил название мутаротации.
Возникновение циклических структур, существование которых предвидел Колли, обусловлено соответственно явлениями d- и g- оксициклотаутомерии.
Для D – ряда a - аномер имеет полуацетальный гидроксил внизу, b - аномер – вверху. Для L – ряда имеет место обратное отношение a - и b - аномеров.
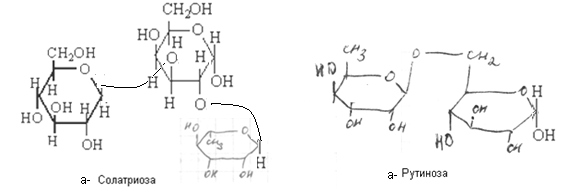
Низкомолекулярные олигосахариды включают в себя также вещества, именуемые «сахара». Это дисахариды (биозы — сложные сахара общей формулы С12Н22О11) и трисахариды (триозы – сложные сахара общей формулы С18Н32О16).
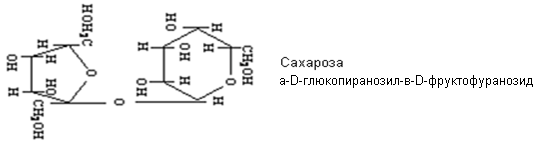
По химической структуре они представляют собой гликозиды, образованные в результате отщепления молекулы воды от двух (биозы) или трёх (триозы) моносахаридов за счет гидроксилов по одному от каждого из них. При этом один из гидроксилов является гликозидным. Если моносахариды все являются способными окисляться (редуцироваться), то среди олигосахаров существуют невосстанавливающие. Если отщепление молекулы воды произошло за счет гликозидных гидроксилов всех молекул моносахаридов, то образуется невосстанавливающие низкомолекулярные олигосахариды. Такие сахара существуют только в циклических конфигурациях и не могут переходить в цепные карбонильные формы и, следовательно, не обладают восстановительными свойствами. В частности, водные растворы их не реагируют с реактивом Фелинга, не подвергаются мутаротации и пр.
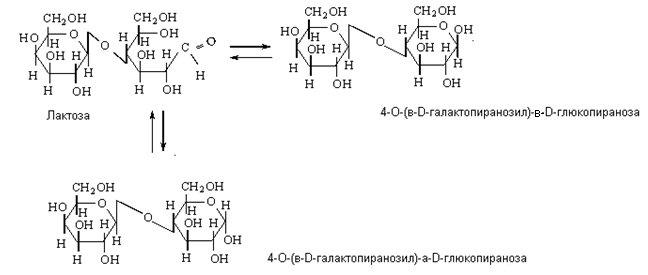
Если отщепление воды произошло за счет гликозидного гидроксила только одного из моносахаридов и негликозидного гидроксила других моносахаридов, то образуется восстанавливающие низкомолекулярные олигосахариды, в котором один из циклов сохраняет гликозидный гидроксил, способный в водных растворах изомеризоваться, превращаясь в цепную карбонильную форму.
Невосстанавливающие олигосахариды
Восстанавливающие олигосахариды
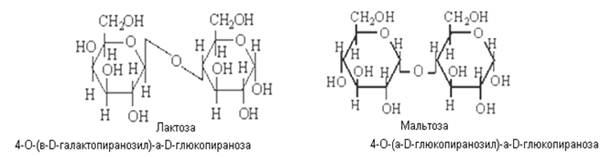
Данным сахарам также присуща мутаротация, и они существуют в a- и b-формах. в циклических и альдегидной таутомерных формах, находящихся в динамическом равновесии:
Гликозидная связь («кислородный» мостик) в молекуле олигосахаров способна очень легко разрываться под действием атаки полярных молекул воды (гидролиз), это весьма важная преданалитическая реакция. Происходит реакция инверсии:
Спирты, содержащие в молекуле свыше трех гидроксильных групп, называют спиртами высшей атомности.
Сахарные спирты или спирты-полиолы являются производными редуцирующих сахаров. По физическим свойствам спирты высшей атомности напоминают редуцирующие сахара — бесцветные, сладкие вещества, образующие сиропообразные растворы.
Химически эти вещества в отличие от сахаров всех видов монофункциональны — не имеют карбонильной группировки, а вместо нее находится группа -СН2ОН (у полиолов, образованных от альдегидоспиртов) или –СНОН (у полиолов, образованных от кетоспиртов).
Таким образом, пентозы образуют спирты-пентиты общей формулы С5Н7(ОН)5 и гексозы —спирты - гекситы общей формулы С6Н8(ОН)6.
Известными представителями спиртов пятиатомных является арабит и ксилит, а наиболее распространенными шестиатомными спиртами являются сорбит, дульцит (галактит) и инозит. Так и называют эти спирты по тривиальной номенклатуре: в названиях соответствующих редуцирующих сахаров заменяют суффикс -оза на -ит. К некоторым имеются также и технические названия. Например:
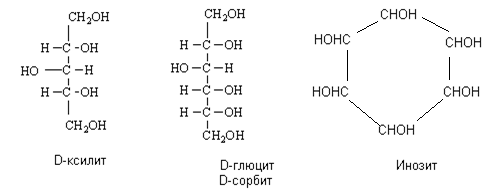
Хотя их чаще всего получают искусственно (реакцией гидрирования на катализаторе никеле) и используют как пищевые добавки-подсластители (ксилит, сорбит), отдельные виды плодов и овощей содержат некоторых сахарных спиртов довольно много. Так, сорбитом богаты ягоды рябины, сок вишен, слив, яблок. Галактит содержится в различных растениях (жасмине, морских водорослях) и в дрожжах и т.д.
В таких сахарах, а также в нередуцирующих олигосахаридах, первостепенное аналитическое значение имеет наличие гидроксильных групп. Не имея карбонильной группы, данные виды сахаров не способны проявлять вышеуказанных свойств редуцирующих сахаров. Атом кислорода в молекуле сахара наиболее электроотрицателен; к нему смещена электронная плотность всех атомов. Следовательно, атом водорода гидроксильной группы имеет большую подвижность, чем атомы водорода в радикале:
![]()
Поэтому в химических реакциях они могут отдавать протон, вступая в частности в реакцию замещения-алкилирования. Алкилированием называют реакции введения в молекулы органических веществ алкильных или сходных по строению радикалов R. При нуклеофильном введении R в молекулу спирта вместо атома водорода ОН-групп получают простые эфиры.
Пентиты содержат два асимметрических атома углерода — С-2 и С-4, третий атом имеет одинаковые заместители СН (ОН)СН2ОН, известны 4 стереоизомера.
Гекситы содержат четыре асимметрических атома углерода, но число стереоизомеров меньше 16, и равно 10 благодаря особенностям строения молекулы. Благодаря наличию одинаковых заместителей у С-2 и С-3, и, соответственно, у С-5 и С-4 молекулы некоторых стереоизомеров имеют плоскость симметрии, находящуюся между атомами С-3 и С-4. Поэтому образовывать оптические стереоизомеры, отклоняя плоскость поляризованного света на поляриметре, могут только те представители, которые не имеют атрибутов симметрии.
Как и моносахариды, они образуют диастереоизомерные ряды (D-рибит и D-арабит).
Химические методы определения сахаров
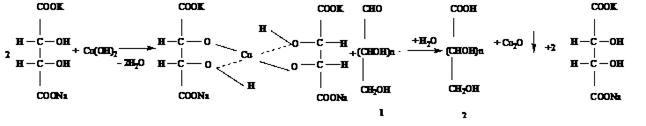
Химические методы разнообразны, однако все они, как и большинство физико-химических, основаны на способности сахаров окисляться в щелочной среде, восстанавливая при этом другие химические вещества с образованием альдоновых кислот. Количество восстановленного другого вещества эквивалентно содержанию сахара в испытуемом растворе. Чаще применяют методы, основанные на окислении сахаров щелочным раствором окисного соединения меди с учетом количества восстановленной меди. Реже применяются методы, в которых используются другие окислители.
Определение восстанавливающих сахаров по методу Бертрана.
Метод Бертрана основан на способности альдегидной группы сахаров взаимодействовать с реактивом Фелинга и восстанавливать окись меди до закиси меди, выпадающей в виде осадка красного цвета:
СuSO4 + 2NaOH ¾¾® Cu(OH)2 + Na2SO4
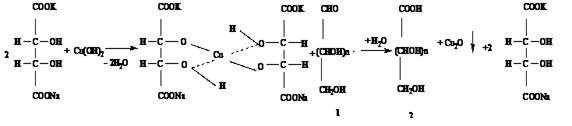
Приведенная реакция не является стехиометрической. Поэтому при пересчете меди на сахар пользуются эмпирическими таблицами, которые составлены при строго определенных условиях протекания реакции.
Реактивы и материалы: а) Реактив Фелинга
20 см3 4 %-ного раствора CuSO4 + 20 см3 щелочного раствора сегнетовой соли {200 г сегнетовой соли растворяют в 500 см3 воды, 150 г едкого натра растворяют в 300 см3. Раствор щелочи осторожно приливают к раствору сегнетовой соли и доводят объем до 1 дм3}.
б) Раствор железоаммиачных квасцов
96 г квасцов растворяют в 500 см3 воды и осторожно по стенке приливают 200 г (108 см3) концентрированной серной кислоты. Объем доводят водой до 1 дм3.
в) 0,1 н. КМпО4
1. Перманганатный метод.
Ход работы:
1.Приготовление вытяжки. Из средней пробы продукта берут навеску, величина которой зависит от предполагаемого содержания сахаров в материале. При исследовании фруктов или ягод навеска составляет 15—50 г мезги (материала, измельченного на терке или мясорубке), варенья, повидла, джема — 7—8 г. При исследовании продуктов, содержащих крахмал (например, клубней картофеля, незрелых яблок и груш), водную вытяжку не нагревают на водяной бане, а сахара извлекают холодной водой в течение 1 ч, часто взбалтывая колбу.
Навеску количественно переносят в мерную колбу на 250 мл, смывая ее дистиллированной водой. Объем навески и воды в колбе не должен превышать 130—150 мл. Колбу встряхивают, затем определяют реакцию содержимого (с помощью нейтральной лакмусовой бумаги или универсального индикатора). При исследовании фруктов и ягод реакция вытяжки обычно бывает кислой, поэтому ее доводят до нейтральной (рН = 7) осторожным добавлением 15%-кого раствора углекислого ратрня (под контролем лакмуса или универсального индикатора), после чего колбу нагревают в течение 15—20 мин, на горячей водяной бане (80°С), часто встряхивая для перемешивания содержимого.
Колбу охлаждают и к вытяжке добавляют 7—15 мл раствора уксуснокислого свинца, взбалтывают и ставят на 5-10 мин. (для осаждения белков, пигментов, дубильных веществ, также обладающих восстанавливающими свойствами). Появление прозрачного слоя жидкости над осадком свидетельствует о полноте осаждения. Если полнота осаждения не была достигнута, в колбу добавляют (каплями) еще 1—5 мл раствора уксуснокислого свинца и взбалтывают. Для осаждения избытка уксуснокислого свинца в колбу приливают 18—20 мл насыщенного раствора двузамещенного фосфорнокислого натрия, взбалтывают и оставляют на 10—12 мин. для отстаивания. Проверяют полноту осаждения свинца, для чего по стенке колбы осторожно приливают 1-—2 капли раствора фосфорнокислого натрия. Если в прозрачном слое жидкости над осадком уже не образуется мути, считают, что полнота осаждения достигнута. Колбу доливают до метки водой, взбалтывают и содержимое ее фильтруют через бумажный складчатый фильтр. В фильтрате (его называют фильтрат А) определяют содержание редуцирующих сахаров. Надо так подобрать навеску продукта и разведение, чтобы концентрация сахаров в сахарном растворе составляла 100 мг.
Быстрого осаждения белковых, красящих и дубильных веществ (так называемых, органических несахаров) можно достигнуть обработкой вытяжки основным азотнокислым свинцом. К 100 мл вытяжки прибавляют 3—4 мл раствора едкого натра, взбалтывают и добавляют 4—6 мл раствора азотнокислого свинца. Осветление раствора происходит в течение 5—7 мин. Для освобождения от избытка свинца к вытяжке, нагретой до температуры 60° С, приливают 3—4 мл насыщенного раствора сернокислого натрия и нагревают на водяной бане при той же температуре 10 мин.
2. Проведение анализа: 20 см3 фильтрата А помещают в коническую колбу на 100 см3 и добавляют 20 см3 реактива Фелинга I и 20 см3 реактива Фелинга II. Содержимое колбы перемешивают и кипятят точно 3 минуты, время замечают с момента появления первых пузырьков. Горячую жидкость из колбы сливают на фильтрующий слой через воронку Бюхнера в колбу Бунзена при слабом отсасывании, стараясь осадок закиси меди не переносить на фильтр. Затем осадок в колбе промывают теплой водой и перерастворяют с помощью железоаммонийных квасцов (10...15см3), при этом часть сернокислого окисного железа квасцов восстанавливается в закисное:
Cu20 + Fe2(NH4)2(SO4)4 + H2SO4 = 2CuSO4 + 2FeSO4 + (NH4)2SO4 + H2O.
Далее воронку Бюхнера с фильтрующим слоем переносят в чистую колбу Бунзена и содержимое колбы небольшими порциями сливают на фильтр. Осадок на фильтре размешивают стеклянной палочкой до полного растворения. Осадок на фильтре не должен находиться на воздухе во избежание его окисления. Колбу и фильтр промывают теплой дистиллированной водой два раза. Фильтрат сразу титруют 0,1 н раствором перманганата калия до появления розовой окраски (от последней капли), снова окисляющего закисное железо в окисное
2KMnO4 + 10FeSO4 + 8H2SO4 = K2SO4 + 2MnSO4 + 5Fe2 (SO4)3 + 8H2O.
Титр перманганата калия устанавливают по меди, что дает возможность сразу пересчитать количество пошедшего на титрование перманганата калия на эквивалентное количество меди (1 см3 0,1 н. КМпО4 соответствует 6,36 мг меди). Количество сахара, соответствующее данному количеству меди, находят по эмпирическим таблицам.
Количество сахара в процентах вычисляют по формуле:
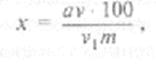
где а — количество сахара в пробе (объем v,); v — объем вытяжки, полученный из навески; vx проба вытяжки (в см3), взятая на определение; т — масса навески материала, мг.
Перманганатный метод считается арбитражным. Существует также ускоренный—йодометрический метод.
2.Иодометрический метод (по Шорлю). Фильтрат А кипятят с жидкостью Фелинга (см. выше). Так как жидкость Фелинга берется в избытке, то часть меди окажется невосстановленной и останется в окисной форме. Чтобы определить избыточное количество окисной меди, в охлажденную после кипячения жидкость добавляют раствор йодистого калия и серной кислоты. Происходит реакция
2 CuSO4 + 4 КJ = Cu2J2 + 2 K2SO4 + I2.
Выделившийся молекулярный иод оттитровывают раствором тиосульфата натрия:
2Na2S2O3 + I2 = Na2S4O6 + 2NaI
Для определения количества двухвалентной меди, восстановленной сахаром, проводят контрольный опыт, в котором вместо исследуемого раствора берут дистиллированную воду. По результату контрольного опыта определяют количество тиосульфата натрия, эквивалентное всей двухвалентной меди, участвующей в опыте. По разности объемов раствора тиосульфата натрия, пошедшего на титрование иода после взаимодействия с KI со всей двухвалентной медью (контрольный опыт) и той, что осталась после взаимодействия с редуцирующими сахарами, судят о количестве восстановленной сахаром двухвалентной меди. Данный метод отличается простотой, высокой точностью определения и возможностью определять содержание сахара в довольно широких пределах (от 0,3 до 88,2 мг в 30 мл раствора).
Реактивы и материалы: Хлеб, нож, разделочная доска, дистиллированная вода, 15%-ный раствор ZnSO4, 4%-ный раствор NaOH, 20%-ный раствор НС1, 10%-ный раствор NaOH, индикатор метиленовый красный, 6,925%-ный раствор CuSO4, щелочной раствор сегнетовой соли, KJ, 25%-ный раствор серной кислоты, 0,1 н. раствор Na2S2O3, 1%-ный раствор растворимого крахмала, мерные колбы вместимостью 100, 200, 250 см3, конические колбы вместимостью 200—300 см3, воронки, цилиндры вместимостью 10 см3, бюретки, водяная баня, термометры, титровальная установка, фарфоровая чашечка вместимостью 20 см3, фильтровальная бумага.
Ход работы:
Проведение анализа: В коническую колбу вместимостью 50 см3 вносят пипеткой 3 см3 фильтрата А, добавляют пипеткой или бюреткой точно 1 см3 6,925%-го раствора сульфата меди (II) и 1 см3 щелочного раствора сегнетовой соли, в течение 2 мин доводят смесь до кипения, кипятят ровно 2 мин, быстро охлаждают до комнатной температуры, прибавляют 1 см3 30 %-ного иодида калия, 1см3 25%-ной серной кислоты и сразу же титруют 0,1 н. раствором тиосульфата натрия до светло-желтого окрашивания. Затем добавляют 3—4 капли 1%-го раствора растворимого крахмала (индикатор) и продолжают титрование до исчезновения синей окраски.
Проведение холостого или контрольного опыта: Аналогично проводят контрольный опыт, в котором вместо 3 см3 исследуемого раствора берут то же количество дистиллированной воды.
Разность между величинами, полученными в контрольном опыте и при определении сахара в исследуемом растворе, умноженная на поправку к титру тиосульфата натрия, показывает количество восстановленной меди, выраженное в см3 точно 0,1 н. раствора тиосульфата натрия. Для пересчета количества 0,1 н. раствора тиосульфата натрия, соответствующего количеству восстановленной меди, на сахар (мг сахарозы) пользуются следующими коэффициентами, установленными экспериментальным путем: глюкоза — 3,3; фруктоза — 3,7; сахароза — 3,4; мальтоза — 5,4.
Наиболее точные показатели получаются в том случае, если разность результатов титрования 0,1 н. раствором тиосульфата натрия в контрольном и основном опытах находится в пределах 0,7—1,2 см3. При использовании вытяжек с более высоким содержанием сахара для определения берут 1 см3 вытяжки и добавляют 2 см3 дистиллированной воды.
3.Определение редуцирующих сахаров (по Лэну и Эй-Нону). В фильтрате А содержатся редуцирующие сахара (глюкоза, фруктоза и другие монозы, а также дисахариды, обладающие восстанавливающими свойствами,— мальтоза, лактоза и др.). Хотя сахароза тоже переходит в фильтрат, но для количественного определения ее необходимо подвергнуть инверсии. Метод определения редуцирующих сахаров основан на титровании реактива Фелинга фильтратом А в присутствии метиленовой сини. Сахара, оставшиеся в небольшом избытке после восстановления окиси меди в закись, реагируют с метиленовой синью, восстанавливая ее в лейкосоединение:
Могут наблюдаться случаи, когда от прибавления метиленовой сини раствор в колбе не посинеет. Это свидетельствует о высокой концентрации редуцирующих сахаров в фильтрате А, и тогда надо его разбавить в два-три раза. Содержание сахаров в испытуемом растворе должно составлять примерно 0,1—0,25%.
Ход работы :
1.Определение титра реактива Фелинга. Титр реактива Фелинга определяют по химически чистой сахарозе. Для установки титра реактива можно также пользоваться сахаром-рафинадом, который предварительно выдерживают в эксикаторе над хлористым кальцием в течение 4—5 суток. На аналитических весах (с точностью до 0,0001 г) отвешивают 0,55 г сахарозы. Навеску переносят в мерную колбу на 250 мл и растворяют в 75 мл теплой воды. К раствору прибавляют 4 мл концентрированной соляной кислоты и производят инверсию сахарозы (в отдельной порции фильтрата А условия инверсии подобраны так, что гидролизуется только одна сахароза). Все последующие операции описаны выше (см. «Определение сахарозы»). Определяют содержание редуцирующих сахаров в растворе. Титр реактива Фелинга (по инвертиому сахару) рассчитывают по формуле
Т = 1,053 ∙ Вгк
Где к — навеска сахарозы, г; В — объем раствора инвертного сахара, израсходованный на титрование 10 мл реактива Фелинга; г — объем раствора инвертного сахара в мерной колос (250 мл); 1,053 —коэффициент перевода сахарозы в инвертный сахар;
2.Проведение анализа:
2.1. Содержание редуцирующих сахаров. В бюретку емкостью 50 мл (со стеклянным краном) наливают фильтрат А, В коническую колбу специальными пипетками вносят по 5 мл растворов Фелинга I и II и вливают из бюретки 15—20 мл фильтрата А. Колбу ставят на электрическую плитку и нагревают (на асбестовой сетке) так, чтобы довести до кипения за 2 мин., после чего прибавляют 4—5 капель раствора метиленовой сини и кипятят точно 2 мин. Продолжая кипячение жидкости, ее титруют из бюретки фильтратом А до исчезновения синего окрашивания и появления оранжевого осадка закиси меди. Титровать надо быстро, чтобы в сумме жидкость кипела не более 3 мин. На дотитровывание следует расходовать не более 2—3 мл фильтрата А. Если при этом расходуется более 3 мл фильтрата А, рекомендуется повторить определение, прибавив в колбу не 15, а уже 20 мл испытуемого раствора. Первое титрование является ориентировочным. Приблизительно установив, сколько миллилитров фильтрата А расходуется на титрование 10 мл реактива Фелинга, проводят два-три точных определения. Содержание редуцирующих сахаров вычисляют по формуле:
Xi = кТ
где Т — титр реактива Фелинга (по инвертному сахару)-; к — навеска растительного материала в объеме испытуемого раствора, израсходованном на титрование 10 мл реактива Фелинга (суммируют количество миллилитров фильтрата А, прибавленных в колбу в самом начале определения и затем затраченных иа дотитровывание) .
2.2. Содержание сахарозы. В мерную колбу на 100 мл вносят 50 мл фильтрата А, добавляют 5 мл концентрированной соляной кислоты (пл. 1,188) и нагревают, часто взбалтывая, в течение 8 мин. на водяной бане, следя за тем, чтобы жидкость в колбе имела температуру 68—70° С (шарик термометра опущен в колбу). Затем колбу быстро охлаждают (под краном) до 20° С. Охлажденную жидкость нейтрализуют углекислым натрием или 15— 20%-ным раствором едкого натра, контролируя этот процесс лакмусовой бумажкой, опущенной в колбу. Нейтрализованную жидкость доводят водой до метки и в случае необходимости фильтруют. Получают фильтрат Б— инвертный сахар — смесь равных частей глюкозы и фруктозы, освободившихся в результате гидролитического расщепления сахарозы. Содержание редуцирующих сахаров в фильтрате определяют по методу, описанному выше. Содержание сахарозы (в процентах) рассчитывают по формуле
Хг = (В - А) 0,95,
где В — содержание редуцирующих сахаров после инверсии; А — то же до инверсии (в процентах); 0,95 коэффициент перевода инвертного сахара в сахарозу.
Суммарное содержание сахаров (в процентах) Х в испытуемом растительном материале рассчитывают по формуле
Х = Xi —1+ Хг 2
где Хi и Хг— содержание соответственно редуцирующих сахаров и сахарозы.
4.Метод горячего титрования. Ускоренный метод, основан на способности редуцирующих сахаров восстанавливать в щелочном растворе окисную медь в закисную. Массовую долю сахара определяют путем титрования медно-щелочного раствора фильтратом А.
Материалы и оборудование: титровальная установка, колбы на 50 мл, реактив Фелинга, электроплитка. Ход работы:
1.Проведение анализа: В бюретку вместимостью 10 см3 наливают исследуемый раствор. В две плоскодонные колбы вместимостью 50 см3 отмеряют пипеткой по 5 см3 раствора Фелинга I и раствора Фелинга II. Одну из колб помещают на нагретую электроплитку, доводят медно-щелочной раствор в колбе до кипения и титруют из бюретки фильтратом А со скоростью (4±1) капель в секунду до перехода синей окраски медно-щелочного раствора в желтую. Израсходованный на титрование объем в см3 стандартного раствора сахарозы отмечают по бюретке.
2.Контрольное титрование. Вторую колбу с медно-щелочным раствором помещают на нагретую электроплитку, раствор в колбе доводят до кипения и сливают в него из бюретки (85±5)% израсходованного на предварительное титрование объема исследуемого раствора, следя за тем, чтобы кипение в колбе не прекращалось. При этом синяя окраска медно-щелочного раствора изменяется на светло-фиолетовую. Дотитрование медно-щелочного раствора исследуемым раствором проводят со скоростью 1 капля в секунду до появления желтой окраски. Массовую долю сахара в исследуемом изделии (М) в пересчете на сухое вещество вычисляют по формуле:
TV1 ∙100 ∙ 2 100 M= ---------------------- ∙ ------------- mV2 ∙ 1000 100 - W
где Т — титр медно-щелочного раствора по сахарозе; V1 вместимость мерной колбы, взятой для приготовления водной вытяжки, см3; m — масса навески исследуемого изделия, г; V2 объем исследуемого раствора, израсходованный на титрование, мл; W массовая доля влаги в исследуемом материале (ГОСТ 21094); 1000 — перевод мг сахарозы в г; 2 — двойное разведение вытяжки при проведении гидролиза сахарозы.
Физико-химические методы определения сахаров
В настоящее время находят широкое применение физико-химические методы определения сахаров. При этом сахара, путем химических реакций, превращают в какое-то вещество, замеряя затем физические характеристики (цвет, адсорбируемость и пр.) Эти методы быстрые, менее трудоемкие, а в некоторых случаях точнее химических.
Количественное определение сахаров в продуктах растительного происхождения с помощью хроматографии на бумаге (по О. А. Павлюшиной)
Количественное определение сахаров с применением хроматографии на бумаге включает в себя следующие основные операции:
фиксацию растительного материала > экстракцию сахаров и очистку вытяжки от белков и других примесей > распределительную хроматографию сахаров на бумаге > элюацию сахаров с бумаги > определение их содержания в элюатах
Реактивы и материалы: а) Хроматографическая бумага Ленинградская «быстрая» № 1; *Filtrak* FN-4; ватман М 1 и 2; б) этанол, 96%-ный и 80%-ный; в) уксуснокислый свинец (СН3СОО)2РЬ ∙ 3Н2О, 10%-ный раствор; г) сернокислый натрий, насыщенный раствор; д) смесь н-бутанола, пиридина и воды в соотношении 6:4:3 (по объему), или смесь н-бутанола, уксусной кислоты и воды в соотношении 4:1:5 (по объему). [Если смеси растворителей разделяются на два слоя, то берут верхний;] е) проявители на кетозы: 1 — резорцинфосфат.
0,2 г резорцина растворяют в 100 мл 96%-ноге этанола. Перед проявлением хроматограммы смешивают 10 объемзв спиртового раствора резорцина с одним объемом ортофосфорной кислоты Н3РО4 (плотность 1,75—1,82);
2— N-хлормочевина.
5 г мочевины растворяют в 100 мл 96%-ного спирта и добавляют 20 мл 2 н раствора соляной кислоты;
ж) проявители на альдозы: 1 — анилинфталат
1,66 г фталевой кислоты и 0,93 г перегнанного анилина растворяют в 100 мл этилового спирта, насыщенного водой;
2 — анилиноксалат:
0,93 г перегнанного анилина растворяют в 50 мл 96%-ного этанола, после чего смешивают с равным объемом 0,2 М раствора щавелевой кислоты в этаноле (можно также брать водный 0,2 М раствор щавелевой кислоты);
и) глюкоза, фруктоза, сахароза и другие сахара, 2%~ные растворы; к) другие реактивы для количественного определения сахаров по антроновому методу.
Ход работы: Навеску свежего растительного материала (10—-30 г) заливают (для фиксации) десятикратным объемом кипящего 96%-ного этанола, нагревают 2—3 мин., добавляя небольшое количество углекислого кальция или натрия в порошке для нейтрализации кислот (под контролем лакмуса или универсального индикатора). Зафиксированная навеска (в колбе или склянке с притертой пробкой) может храниться в течение нескольких месяцев (в темном и прохладном месте). Фиксация спиртом является и началом экстракции сахаров, которые частично переходят в раствор. Сахара экстрагируют горячим (70—80 ºС) 80%-ным этанолом 3 раза по 30 мин. Перед началом экстракции рекомендуется дополнительно растереть материал в ступке. Спирт, который служил для фиксации навески, объединяют со спиртовыми вытяжками. После каждой экстракции охлажденную спиртовую вытяжку центрифугируют. Объединенные спиртовые вытяжки сгущают в вакууме до объема 3 мл. Если материал богат хлорофиллом и каротиноидами, то сгущенную вытяжку несколько раз взбалтывают с петролейным эфиром. Эфирный раствор пигментов сливают, а остатки растворителя удаляют на водяной бане при 50—60°. Сгущенную вытяжку количественно переводят в мерную колбу на 5 или 10 мл. Для осаждения белков и других примесей прибавляют по каплям раствор уксуснокислого свинца. Проверив полноту осаждения, раствор в колбе доводят до метки, затем фильтруют или, еще лучше, центрифугируют. Для осаждения избытки свинца, не пошедшего в реакцию, добавляют одну каплю насыщенного раствора сернокислого натрия и снова центрифугируют или фильтруют. Нарезают полосы хроматографической бумаги длиной 50—55 см и шириной 13—19 см или другого размера (в зависимости от диаметра хроматографической камеры). Бумагу разлиновывают графитовым карандашом, оставляя с одной стороны листа одну, а с другой — две контрольные полосы шириной 2— 2,5 см. Низ хроматограммы вырезают зубцами, что способствует равномерному скапыванию растворителя и предотвращает наблюдающийся иногда перекос пятен. Затем на линию старта отдельными пятнами по одному на основной части хроматограммы и на каждой из контрольных полос наносят с помощью микропипетки или капилляра определенный точный объем испытуемого раствора (например, по 0,01; 0,02 или 0,03 мл в пятно). Количество экстракта, наносимого в пятно, должно соответствовать примерно 30—50 мг сырого веса исследуемого материала в том случае, если в нем содержится 5—7 мг глюкозы, фруктозы и сахарозы на 1 г сырого веса ткани. Для установления оптимальной концентрации сахаров, вносимых в пятно, ставят отдельно две контрольные хроматограммы с нанесением различных объемов опытного экстракта (0,01—0,04 мл в пятно). На те же полосы наносят также и метчики — растворы сахаров (по 0,004 мл 2%-ных растворов). После нанесения растворов на бумагу приступают к разделению сахаров в соответствующей смеси растворителей (н-бутанол — пиридин — вода, 6:4:3; н-бутанол — уксусная кислота — вода, 4:1:5). Хроматография нисходящая. Продолжительность разделения 2—3 суток. После разделения хроматограммы высушивают в токе воздуха (в вытяжном шкафу) до полного удаления следов растворителя. Следующей задачей является определение положения пятен на основной части хроматограммы, так как сахара элюируют из непроявленной части бумаги. Для этого от основной хроматограммы отрезают контрольные полосы (а), (б) и (в) и опрыскивают две из них, например (а) и (б), проявителями на кетозы — резорцинфосфатом (реакцией Ф.Ф. Селиванова):
или раствором N-хлормочевины:
а (в) — проявителем на альдозы — анилинфталатом (анилиноксалатом):
Полосу, опрыснутую резорцинфосфатом, прогревают 3—4 мин. в сушильном шкафу при температуре 85—95ºС. Пятна фруктозы, сахарозы, раффинозы и олигосахаридов, содержащих фруктозу, окрашиваются в интенсивно розовый цвет. При нагревании хроматограммы, опрыснутой раствором хлормочевины, в течение 5 мин. при 105 ºС пятна альдоз принимают синее окрашивание. Полосу, обработанную раствором анилинфталата или анилиноксалата, нагревают при 105—110 ºС. При обработке хроматограммы анилинфталатом глюкоза и галактоза проявляются в течение 5 мин. при 105°С в виде желтовато-коричневых пятен; пятна пентоз вишнево-красного цвета. Мальтоза и лактоза дают желто-коричневые пятна при нагревании в течение 20 мин. при 105 ºС. При проявлении анилиноксалатом (110°С, 5—6 мин.) пятна альдоз имеют коричневое окрашивание. Проявленные контрольные полосы (а), (б) и (в) прикладывают к основной части хроматограммы (в) и таким образом определяют положение пятен отдельных сахаров на полосе (в), не проявляя ее. Участки бумаги, содержащие не проявленные пятна глюкозы, фруктозы, сахарозы и олигосахаридов, вырезают, разрезают на маленькие кусочки («лапшу») и элюируют сахара водой три раза по 30 мин. при 60—70°. Элюаты фильтруют через маленькие бумажные фильтры, выпаривают на водяной бане до небольшого объема, снова фильтруют и доводят до определенного объема. В растворе определяют содержание сахара с помощью антронового реактива.
Определение восстанавливающих сахаров колориметрическим методом (по И. С. Лурье)
Метод основан на взаимодействии восстанавливающих сахаров при нагревании со стандартным щелочным раствором красной кровяной соли. При этом ее часть восстанавливается в желтую кровяную соль.
С6Н12О6 + 2K3[Fe(CN)6] + 2KOH = СН2ОН(СНОН)4СООН + 2K4[Fe(CN)6] + Н2О
Избыток феррицианида определяют на фотоэлектроколориметре по характерному поглощению в области 420...440 нм (синий светофильтр). Расчет восстанавливающих сахаров ведут по калибровочной кривой. Метод удобен для серийных анализов и применим в тех случаях, когда испытуемый раствор не содержит других веществ, взаимодействующих с феррицианидом, а также веществ, имеющих поглощение в данной области спектра. При работе с биологическим материалом, исследуемые вытяжки бывают очень сложны по своему составу, часто опалесцируют, поэтому требуется определенная подготовка образца. Метод очень удобен для изучения накопления восстанавливающих сахаров под действием ферментов, гидролизующих углеводы, в экспериментах, моделирующих технологический процесс. В этом случае исследователя интересует не абсолютное содержание восстанавливающих сахаров, а их увеличение за счет ферментативной реакции. При этом факторы, влияющие на величину оптической плотности, нивелируются контрольными или нулевыми замерами.
Реактивы и материалы: а) 1 %-ный раствор феррицианида (10 г в 1 дм3). б) 1,25 н. раствор КОН (70 г в 1 дм3). в) 1 %-ный исходный раствор глюкозы. г) 0,1 %-ный и 0,2 %-ный рабочие растворы глюкозы.
Ход работы.
1.Выполнение анализа: В коническую колбу на 100 см3 вносят 10 см3 раствора феррицианида, 5 см3 раствора щелочи и от 1 до 5 см3 испытуемого раствора. Если объем пробы меньше 5 см3, то недостающее количество компенсируют водой. Колбочку прикрывают часовым стеклом и на слабом огне доводят до кипения. Кипятят ровно 1 минуту. Затем содержимое колбочки охлаждают до комнатной температуры и измеряют оптическую плотность на ФЭКе при синем светофильтре (420 нм). Количество сахара в испытуемом растворе определяют по калибровочной кривой.
2.Построение калибровочной кривой: Концентрацию исходного раствора глюкозы устанавливают по методу Бертрана. Для этого берут три пробы по 5 см3 1 %-ного раствора глюкозы, добавляют к каждой пробе по 15 см3 воды и 40 см3 реактива Феллинга. Концентрацию исходного раствора глюкозы рассчитывают как среднее значение из трех параллельных определений. Точную концентрацию рабочих растворов глюкозы рассчитывают по данным анализа исходного раствора. Сухие конические колбочки на 100 см3 заполняют согласно табл. 2.
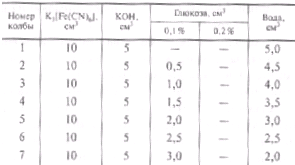


В заполненных колбочках проводят определение содержания глюкозы колориметрическим методом и строят калибровочную кривую в координатах «оптическая плотность — количество глюкозы, мг».
Определение восстанавливающих сахаров методом Шомадьи — Нельсона (предшественник метода Фолина-Ву)
Колориметрический метод Шомадьи — Нельсона основан на реакции восстанавливающих сахаров с реактивом Шомадьи — Нельсона, в результате которой образуется окрашенное соединение «молибденова синь» общего состава MonO3n-x голубого цвета. Интенсивность окраски пропорциональна содержанию восстанавливающих углеводов, которые определяют по величинам оптических плотностей при 508 нм, по калибровочной кривой, составленной с применением модельных растворов. Метод неспецифичен, трудоемок.
Реактивы и материалы: а) Реактив Шомадьи.
a 24 г карбоната натрия и 12 г тартрата калия — натрия (калий натрий виннокислый средний) помещают в коническую колбу вместимостью 500 см3 и добавляют 250 см3 дистиллированной воды. Затем приливают 40 см310 % раствора сульфата меди, перемешивают и добавляют 16 г гидрокарбоната натрия.
b.18 г Na2SO4 растворяют в 500 см3 горячей дистиллированной воды. Раствор кипятят в течение 40 минут для удаления СО2, затем охлаждают.
Cмешивают (a+b) в мерной колбе на 1 дм3, доводят до метки дистиллированной водой и перемешивают. В течение первых двух-трех суток хранения реактива Шомадьи оседает небольшое количество меди с примесями. Этот осадок отфильтровывают, готовый раствор хранят в темной склянке.
б) Реактив Нельсона.
a 25 г модибдата аммония помещают в мерную колбу на 500 см3 и растворяют в 150 см3 Н2О. Раствор перемешивают, к нему осторожно небольшими порциями добавляют 21 см3 концентрированной H2SO4 (х. ч.) при непрерывном перемешивании.
b 3 г гептагидрата гидроортоарсената натрия (Na2HAsO4 7Н2О) растворяют отдельно в химическом стаканчике в 50 см3 Н2О
(a+b). Получается прозрачный раствор светло-желтого цвета, его доводят до метки водой и выдерживают в термостате при 37 "С в течение 2 суток. После выдержки раствор готов к употреблению.
Ход работы.
1.Проведение анализа: К 1 см3 испытуемого раствора добавляют 1 см3 реактива Шомадьи, перемешивают и выдерживают на кипящей водяной бане в течение 15 минут:
СuSO4 + 2NaOH [гидролитический] ¾¾® Cu(OH)2 + Na2SO4
Затем к охлажденной смеси добавляют 1 см3 реактива Нельсона и доводят объем до 10 см3 дистиллированной водой:
Na2HAsO4 + 15H2SO4 + 12(NH4)2MoO4 [в реактиве Нельсона]¾¾® (NH4)3H4[As(Mo2O7)6] + 21(NH4)2SO4 + Na2SO4 + 10H2O
2(NH4)3H4[As(Mo2O7)6] + Cu2O ¾¾® 4Mo6O17 + 2CuO +(NH4)2SO4 + 2(NH4)2HAsO4 + 3H2O
Оптическую плотность полученного раствора измеряют при 590 нм с использованием кюветы с шириной грани 5 мм.
2.Построение калибровочной кривой. Калибровочную кривую строят после проведения колориметрической реакции модельных растворов глюкозы с концентрацией от 0,02 до 0,14 мг/см3. Изучаемая зависимость выражается прямой линией, выходящей из начала координат.
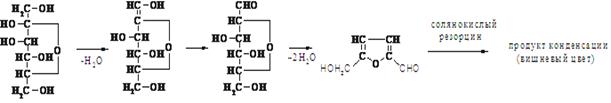
Определение фруктозы и других кетосахаров (по Мак-Рери и Слаттери)
Определение основано на способности кетосахаров давать вишневую окраску с резорцином в кислой среде:
Реактивы и материалы: а) Спиртовой раствор резорцина
1 г резорцина растворить в 1 дм3 95%-ного этанола.
б) 30 %-ный раствор НС1 (не должен давать окраску с резорцином).
в) Стандартный раствор фруктозы
100 мг фруктозы растворяют в 100 см3 насыщенного водяного раствора бензойной кислоты.
г) Рабочий раствор фруктозы
1 см3 стандартного раствора фруктозы разводят в мерной колбе на 100 см3
Ход работы. В пробирку вносят 5 см3 испытуемого раствора (содержащего от 1 до 8 мг фруктозы в 100 см3 раствора), 5 см3 спиртового раствора резорцина и 15 см3 30 %-ного раствора НСI. Содержимое пробирки перемешивают и помещают на 20 минут в водяную баню при температуре 80 °С. Затем содержимое пробирки охлаждают до комнатной температуры и измеряют оптическую плотность на ФЭКе при 540 нм (зеленый светофильтр). В качестве контроля используют раствор, в котором вместо 5 см3 испытуемого раствора берется 5 см3 Н2О. Количество фруктозы определяют по калибровочной кривой. Для этого готовят серию разведений рабочего раствора фруктозы, в каждом из которых проводят определение фруктозы по описанной выше методике. Калибровочную кривую строят в координатах: «оптическая плотность — количество фруктозы, мг».
Газохроматографическое определение отдельных сахаров
Метод основан на переводе углеводов типа глюкозы, фруктозы, арабинозы, ксилозы, галактозы, сахарозы, мальтозы, лактозы, раффинозы, а также полиолов — сорбита и инозита в пищевых продуктах в триметилсилильные производные с последующей их идентификацией на газовом хроматографе.
Аппаратура, реактивы и материалы: а) Газовый хроматограф с пламенно-ионизационным детектором и устройством для программирования температуры. б) Стеклянная насадочная колонка длиной 2...3 м и диаметром З...4 мм или капиллярная колонка длиной 25...30 м с нанесенной фазой. в) Микрошприц емкостью 1,0...10мкл. г) Роторный испаритель. д)Гексан, х.ч. е)Гексаметилдисилазан. ж) Трифторуксусная кислота или триметилхлорсилан. з) Пиридин, х.ч., безводный. и) Ксилит, х.ч. или инозит. й) Свинец уксуснокислый, х.ч. к) Неподвижные фазы для ГЖХ SE-30, OV-17, ХЕ-60, СКТФТ-50. л) Инертный носитель для ГЖХ: хромосорб-W DMCS, хроматон-N DMCS.
Ход работы.
1. Подготовка образцов к анализу углеводов. Для получения достоверных результатов анализа необходимо учитывать содержание жира и лимонной кислоты в испытуемом образце. Предварительно содержание этих компонентов в пищевых продуктах можно оценить по «Таблицам химического состава пищевых продуктов» и на основании этого выбрать один из следующих способов подготовки образца к анализу.
1.1. Продукты с низким содержанием жира (менее 1,5%).
1.1.1.
Продукты с низким содержанием лимонной кислоты (менее 1,5 %).
Содержание лимонной кислоты устанавливается по «Таблицам химического состава».
К ним относятся соки, вина, пиво, фруктово-ягодные кондитерские изделия, мед,
сиропы и др. Навеску гомогенизированной пробы продукта с высоким
содержанием сухих веществ (более 60 %) наносят на полиэтиленовую пластинку в
количестве 50... 150 мг и взвешивают на аналитических весах с точностью 0,0001
г. На этой же пластинке взвешивают 5... 10 мг ксилита. Пластинку помещают в
круглодонную колбу емкостью 10...25 см3 и силилируют без
обезвоживания с использованием трифторуксусной кислоты. При использовании
триметилхлорсилана содержимое колбы упаривают досуха на роторном испарителе при
60 "С под вакуумом. В случае медленного упаривания в колбу добавляют 1 см3
бензола. После упаривания проводят силилирование. ![]() Жидкие
продукты отмеряют пипеткой в количестве 0,5...
1,0 см3 и помещают в круглодонную колбу емкостью 10...25 см3,
снабженную шлифом. Туда же на полиэтиленовой пластинке вносят навеску 5... 10
мг ксилита, взвешенного с точностью 0,0001 г. Содержимое колбы упаривают досуха
на роторном испарителе.
Жидкие
продукты отмеряют пипеткой в количестве 0,5...
1,0 см3 и помещают в круглодонную колбу емкостью 10...25 см3,
снабженную шлифом. Туда же на полиэтиленовой пластинке вносят навеску 5... 10
мг ксилита, взвешенного с точностью 0,0001 г. Содержимое колбы упаривают досуха
на роторном испарителе.
1.1.2. Продукты с содержанием лимонной кислоты более 1,5 %. Для исключения наложения пика ТМС-производного лимонной кислоты на ТМС-производное (3-глюкозы, лимонную кислоту осаждают уксуснокислым свинцом. Навеску средней гомогенизированной пробы продукта (свежие цитрусовые плоды, цитрусовые соки, продукты, содержащие лимонную кислоту как пищевую или консервирующую добавку) в количестве 1...1,5г взвешивают с точностью до 0,0001 г в конической колбе емкостью 100 см3, приливают 30 см3 75...80 %-ного раствора этанола и экстрагируют 30 минут при температуре 60...70°С. Используют водяную баню и обратный холодильник. Экстракцию повторяют трижды, экстракты объединяют. Содержимое колбы доводят до метки дистиллированной водой. В центрифужную пробирку на 10 см3 вносят пипеткой 5 см3 отфильтрованного экстракта, 0,5 см3 насыщенного раствора уксуснокислого свинца. Осадок центрифугируют. 1 см3 надосадочной жидкости переносят в круглодонную колбу со шлифом емкостью 10...25 см3. Туда же на полиэтиленовой пластинке вносят навеску ксилита 5... 10 мг. Содержимое колбы упаривают на роторном испарителе при 60 "С под вакуумом.
1.2. Продукты с высоким содержанием жира (более 1,5%).
1.2.1. Продукты с содержанием лимонной кислоты менее 1,5 % (детское молочное питание, сухие молочные смеси и др.). Навеску средней гомогенизированной пробы продукта с высоким содержанием сухих веществ (более 60%) в количестве 500...1000 мг помещают в центрифужную пробирку. Туда же на полиэтиленовой пластинке вносят с точностью до 0,0001 г навеску ксилита 2...5 мг. Проводят экстракцию с 2 см3 гексана с последующим центрифугированием. Экстракцию проводят трижды, декантируя гексановый слой. Остатки гексана высушиваются в токе азота, после чего проводят силилирование.
Пробу сгущенного молока взвешивают в центрифужной пробирке и проводят обезжиривание, как описано выше, затем добавляют 20 см3 50 %-ного водного раствора этанола, тщательно перемешивают, затем обрабатывают аналогично жидким молочным продуктам.
10 см3 жидких молочных продуктов помещают в центрифужную пробирку, смешивают с 10 см3 96 %-ного этанола, выдерживают 30...40 минут и центрифугируют при 4000 об/мин в течение 10 минут. 1 см3 надосадочной жидкости помещают в круглодонную колбу, туда же на полиэтиленовой пластинке вносят 2...5 мг ксилита, содержимое упаривают насухо на роторном испарителе при 60 °С, после чего проводят силилирование.
1.2.2. Продукты с содержанием лимонной кислоты более 1,5 % и хлебобулочные изделия. Навеску средней гомогенизированной пробы продукта в количестве 10...15 г, взвешенную с точностью до 0,0001 г, помещают в центрифужную пробирку и подвергают трижды экстракции 30 см3 гексана, объединяя гексановый слой декантированием. Осадок шпателем переносят в коническую колбу вместимостью 100 см3, тщательно смывая остатки со шпателя и пробирки 75 %-ным раствором этанола. Затем трижды экстрагируют содержимое колбы 75 %-ным раствором этанола, подогревая колбу на водяной бане при 60 °С. Отфильтрованные экстракты объединяют и доводят дистиллированной водой до метки в мерной колбе на 100 см3. В мерную пробирку на 10 см3 вносят 5 см3 экстракта и 0,5 см3 насыщенного раствора уксуснокислого свинца. После выпадения осадка надосадочную жидкость в количестве 1,0 см3 переносят в круглодонную колбу вместимостью 10...25 см3. Туда же вносят на полиэтиленовой пластинке 2...5 мг ксилита (с точностью до 0,0001 г) и содержимое колбы упаривают на роторном испарителе при 60 °С под вакуумом, после чего проводят силилирование.
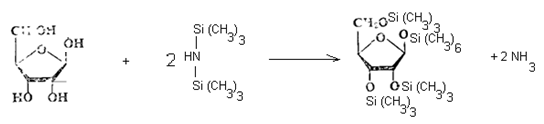
2. Силилирование сахаров.
2.1. Способ с использованием трифторуксусной кислоты. В подготовленную навеску образца приливают 1,0 см3 пиридина (нуклеофильный катализатор), 0,9 см3 гексаметилдисилазана, 0,1 см3 трифторуксусной кислоты (облегчающий алкилирование), плотно закрывают и энергично встряхивают в течение 30 секунд. Вначале наблюдают расслоение жидкости на 2 фазы, при этом нижний слой незначителен. По мере стояния раствора в течение 20...30 минут это расслоение исчезает и начинает выделяться аммиак, что указывает на успешное протекание реакции силилирования:
После прекращения выделения аммиака раствор выдерживают 12 часов при комнатной температуре или 1 час при 60 "С. Длительно сохраняющееся расслоение, исчезающее только при нагревании, говорит о том, что реакция силилирования прошла не полностью из-за высокого содержания влаги (более 40%) или повышенного содержания углеводов. В этом случае подготовку пробы повторяют, уменьшив при этом навеску или увеличив время высушивания на роторном испарителе.
![]()
![]() 2.2. Способ с
использованием триметилхлорсилана. В подготовленную
навеску образца приливают точно 1,0 см3 пиридина, 0,2 см3
гексаметилдисилазана и 0,1 см3 триметилхлорсилана, встряхивают в
течение 1 минуты и нагревают в термостате 45 минут при 60 °С:
2.2. Способ с
использованием триметилхлорсилана. В подготовленную
навеску образца приливают точно 1,0 см3 пиридина, 0,2 см3
гексаметилдисилазана и 0,1 см3 триметилхлорсилана, встряхивают в
течение 1 минуты и нагревают в термостате 45 минут при 60 °С:
Затем хроматографируют.
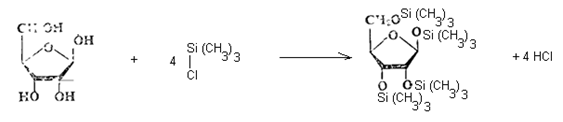
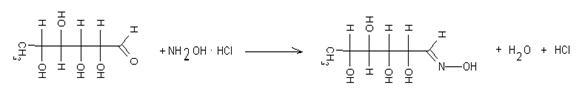
Для упрощения идентификации и количественных расчетов (если не требуется знания аномерного состава сахаров) определение сахаров производят в виде ТМС-производных сахаров. В подготовленную пробу образца добавляют 100 г гидроксиламина солянокислого, приливают 10 см3 пиридина и выдерживают в термостате при 80 °С в течение 2 часов:
Охлаждают и далее приливают силилирующие агенты.
3. Газохроматографический анализ.
3.1.Подготовка хроматографической колонки. Стеклянную колонку, заполненную готовым сорбентом (количество неподвижной фазы 3...5% от массы носителя) длиной 2 м и внутренним диаметром З мм устанавливают в термостате хроматографа и проводят термокондиционирование при продувании потоком газа-носителя (гелий, азот, водород) 2 ч при 100°С, 2 ч при 150°С, 4 ч при 200°С и 8 ч при 250"С. Затем устанавливают рабочий режим: температуру колонки профаммируют от 125 до 270°С со скоростью 4 °С в 1 мин, температуру испарителя 250 °С, расход газа-носителя 40см3/мин, температура пламенно-ионизационного детектора 250 °С, продолжают кондиционировать в течение рабочего дня.
3.2.Газохроматографическое определение. 1 мкл пиридинового раствора триметилсилильных производных углеводов вводят в испаритель и элюируют из колонки газом-носителем. Идентификацию индивидуальных триметилсилильных производных проводят по времени удерживания триметилсилильных производных сахаров-метчиков и методом добавок.
3.3. Приготовление стандартных растворов сахаров.
3.3.1. Сахара, имеющие аномеры . Навеску ксилита и навеску определяемого сахара, взятую с точностью 0,0001 г, помещают в коническую колбу и заливают дистиллированной водой до полного растворения углеводов. Раствор выдерживают в течение суток. Аликвотную часть раствора отбирают пипеткой, помещают в круглодонную колбу и упаривают досуха, после чего проводят силилирование.
3.3.2. Сахара, не имеющие аномеров. Силилируются без предварительного растворения.
Массовую долю отдельных сахаров (в %) в навеске продукта определяют по формуле:
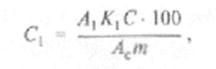
где С, — содержание отдельного сахара в навеске, %; К1 — поправочный коэффициент данного сахара; С — масса навески стандарта, мг; m масса навески образца; Ас — площадь пика стандарта в относительных единицах; Лс— площадь пика данного сахара в относительных единицах.
Ввиду того, что альфа-лактоза и сахароза выходят на хроматограмме одним пиком, площадь пика альфа-лактозы определяют, исходя из соотношения площадей альфа- и бета-лактозы в модельном соединении лактозы. За основу берется площадь пика бета-лактозы. Площадь пика сахарозы определяют вычитанием от суммарного пика сахарозы пика альфа-лактозы, из пика бета-лактозы. За окончательный результат принимают среднеарифметическое результатов двух параллельных определений и округляют до трехзначной цифры.
Количественное определение сахаров по реакции с пикриновой кислотой (по Крезелиус – Зейферт)
При взаимодействии редуцирующих сахаров с пикриновой кислотой они окисляются до соответствующих кислот, а пикриновая кислота восстанавливается в пикраминовую, обладающую красной или буровато-красной окраской:
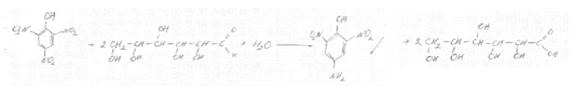
Метод пригоден для количественного определения глюкозы, галактозы, арабинозы, фруктозы, рамнозы, ксилозы, мальтозы, лактозы и крахмала. Метод быстр, но не очень точен. Ошибка может превышать 10-20%. В связи с этим указанный метод имеет ориентировочное значение;
Реактивы и приборы: а) пикриновая кислота, насыщенный раствор; б) углекислый натрий, 20%-ный раствор; в) спектрофотометр.
Ход работы: К 1 мл испытуемого раствора (в котором должно содержаться ее более 3 мг редуцирующих сахаров) прибавляют 2 мл насыщенного водного раствора пикриновой кислоты и 1 мл раствора углекислого натрия, после чего пробирку переносят на 30 мин. в кипящую водяную баню. Охлаждают до комнатной температуры и доводят дистиллированной водой до объема 10 мл. Оптическую плотность раствора определяют при 455 нм (синий светофильтр). Содержание редуцирующих сахаров рассчитывают по калибровочной кривой, составленной по стандартным растворам глюкозы как в работе «Количественное определение сахаров по антроновому методу».
Определение редуцирующего общего сахара по реакции с 3,5-дииитросалицнловой кислотой
При взаимодействии сахаров с 3,5-динитросалициловой кислотой она восстанавливается в З-амино-5-нитросалицнловую:
В остальном реакция протекает так же, как и с пикриновой кислотой.
Реактивы и приборы: а) динитросалициловый реактив:
В мерной колбе на 1 л растворяют в дистиллированной воде 10 г 3,5-динитросалициловой кислоты, 300 г сегнетовой соли, 16 г едкого натра и доводят водой до метки. Раствор выдерживают два дня в темном месте, затем фильтруют в склянку оранжевого стекла и хранят в темноте;
б) спектроколориметр «Спекол» (ГДР).
Ход работы: К 1 мл испытуемого раствора (где не более 2 мг редуцирующих сахаров) прибавляют 2 мл динитросалицилового реактива, нагревают 5 мин. в кипящей водяной бане, охлаждают до комнатной температуры, после чего доводят до объема 25 мл. Оптическую плотность раствора определяют при 530 нм (зеленый светофильтр).
Антроновый метод определения сахаров (по Моррису-Роэ)
Антроновый реактив образует зеленое окрашивание со всеми растворимыми углеводами, которые в одинаковой концентрации дают окрашенные растворы практически одной и той же оптической плотности.
Это позволяет при определении углеводов использовать калибровочную кривую, составленную по глюкозе, для определения других сахаров в небольших концентрациях (до 0,2 мг в пробе). Точные результаты могут быть получены при наличии высокоочищенных химических реактивов и соблюдении постоянной температуры.
Реактивы и материалы: а) Антроновый реактив
200 мг антрона растворяют в 100 см3 концентрированной серной кислоты (плотность 1,84).
б) 0,5%-ный раствор H2SO4
2,8 см3 серной кислоты, плотностью 1,84 растворяют в 1 дм3 дистиллированной воды.
в) 30 %-ный раствор сернокислого цинка г)15 %-ный раствор желтой кровяной соли 3-водной.
Ход
работы. 1 г измельченного
материала (корнеплодов, плодов и др.) помещают в 2 мерные колбы на 100 см3.
Проводят извлечение сахаров в течение часа с 80 см3 воды при
периодическом перемешивании. После этого экстракт осветляют, прибавляя по 1 см3
растворов сернокислого цинка и желтой кровяной соли. Берут 2 пробирки и
приливают по 3 см3 антронового реактива, а затем отбирают 1 см3
опытной вытяжки. В качестве контроля используют пробирки, в которых вместо
вытяжки добавляют 1 см3 воды. Все пробирки быстро взбалтывают
и помещают в кипящую водяную баню на 7 минут. После кипячения пробирки с
растворами охлаждают до комнатной температуры, определяют оптическую плот![]()
![]()
![]()
![]() ность при 610 нм
против контрольной пробы. По калибровочной кривой рассчитывают содержание сахаров.
Расчет производят по формуле:
ность при 610 нм
против контрольной пробы. По калибровочной кривой рассчитывают содержание сахаров.
Расчет производят по формуле:
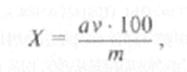
где а — количество сахаров, найденное по калибровочной кривой; v объем экстракта (см3); т — масса навески, взятой на определение.
Определение содержания общего сахара в продуктах кондитерского производства
Считается экспресс-методом, основанным на окислении сахаров дихроматом калия в сильнокислой среде по общей схеме:
![]()
Соединения хрома (Ш) окрашены в сине-зеленый цвет, их количество пропорционально содержанию общего сахара в анализируемом продукте.
Реактивы и материалы:
а) Раствор сахарозы 0,004 г/см3 (стандартный раствор сахарозы).
1,0000 г сахарозы взвешивают с точностью до 0,0002 г и растворяют в мерной колбе вместимостью 250 см3. Объем раствора доводят до метки дистиллированной водой.
б) 1 %-ный раствор дихромата калия (основной реактив).
49 г дихромата калия растворяют при нагревании в 300см3 воды, отдельно в 300 см воды медленно при перемешивании вводят 300 см3 концентрированной H2SO4 и охлаждают. В мерную колбу вместимостью 1000 см3 сначала помещают раствор дихромата калия, затем — серную кислоту, объем раствора доводят водой до метки, осторожно перемешивают.
в) Серная кислота плотность 1,84 г/см3. г) 0,5 М раствор сульфата цинка. д) 1 М раствор гидроксида натрия.
Ход работы.
1. Подготовка к анализу
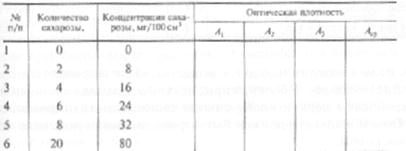
1.1. Построение градуировочного графика. В 6 мерных колб вместимостью 100 мл вносят по 25 см раствора дихромата калия. Из бюретки последовательно добавляют 0; 2; 4; 6; 8 и 20 см3 стандартного раствора сахарозы. Затем во все колбы из бюретки приливают дистиллированную воду до объема 50 см3, (т. е. добавляют 25, 23, 21, 19, 17 и 5 см3 воды). Получают серии растворов, содержащих соответственно 0, 8, 16, 24, 32 и 80 мг сахарозы в 100 см3. Содержимое колбы нагревают на кипящей водяной бане 10 мин, охлаждают под струей водопроводной воды, объем растворов доводят до метки дистиллированной водой и перемешивают. Измеряют оптическую плотность полученных растворов на фотоэлектроколориметре при длине волны 670 нм и толщине кюветы 5 см. Раствором сравнения является раствор с нулевой концентрацией сахарозы. Оптическую плотность определяют в каждом растворе не менее 3 раз. Для построения калибровочного графика в координатах «оптическая плотность — количество сахарозы, мг/100см3» используют среднее значение оптической плотности согласно таблице 3:
1.2. Приготовление вытяжки. Анализируемый образец измельчают в ступке. Затем готовят водную вытяжку объекта исследования. Примерные навески для различных пищевых объектов приведены в таблице 4:
Навеску с точностью до 0,01 г переносят в мерную колбу вместимостью, определенной по табл. 2.5 в зависимости от взятой пробы. Навеску растворяют в дистиллированной воде, нагретой до 60 град С: А. Если изделие растворяется в воде без остатка (сахарные леденцы, сиропы и др.), то полученный в стаканчике раствор охлаждают и переносят в мерную колбу. Объем раствора доводят до метки дистиллированной водой и хорошо перемешивают.
1.3.Отделение мешающих несахаров: Если в изделии находятся вещества, нерастворимые в воде «мешающие несахара» — белки, жиры, пектины, крахмал и т. п., то навеску переносят в мерную колбу, смывая частицы дистиллированной водой. Объем жидкости должен быть примерно равен половине объема мерной колбы. Колбу с жидкостью помещают на водяную баню, нагретую до 60 °С (для объектов, содержащих крахмал до 50 °С). При этой температуре выдерживают колбу в течение 15 мин, периодически перемешивая. За это время практически все сахара переходят в раствор. Охладив раствор, осаждают «мешающие несахара», прибавляя к нему 5см3 0,5 М раствора ZnSO4 и 2,5 см3 1 М раствора КОН или NaOH. Раствор доводят до метки и фильтруют.
2. Проведение анализа. В мерную колбу вместимостью 100 см3 отбирают цилиндром 25 см3 раствора дихромата калия, 10 см3 прозрачного фильтрата и 15 см3 воды, нагревают в течение 10 мин на кипящей водяной бане, охлаждают, добавляют до метки воду, перемешивают. Полученным раствором заполняют кювету и определяют оптическую плотность также, как и при снятии градуировочного графика. По градуировочному графику находят содержание сахарозы (мг/100см3) раствора, что соответствует содержанию сахарозы во взятой пробе, выраженному в мг.
Содержание общего сахара X (%) в анализируемом продукте вычисляют по формуле:
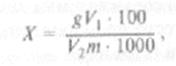
где g — содержание общего сахара, найденное по градуировочному графику, мг/100 мл; V, вместимость мерной колбы, мл; V2 объем фильтрата, взятый для реакции с дихроматом калия, мл; т масса навески объекта исследования, г.
Физические методы определения сахаров
Известно не очень много таких методов. Физические методы определения сахаров основаны на измерении явственных физических свойств сахаров специальными приборами, градуированными по корреляции «концентрация раствора — сила физического свойства раствора». Достоинства: простота, быстрота, отсутствие дорогостоящих реактивов и химических превращений. Недостатки: не слишком высокая воспроизводимость результатов.
Рефрактометрический метод
Принцип действия рефрактометра основан на явлении полного внутреннего отражения при прохождении светом границы раздела двух сред с разными показателями преломления. Измерения проводят при дневном свете, или при включенном осветителе в проходящем через прозрачную исследуемую среду свете, или в отраженном свете, когда исследуемая среда существенно поглощает или рассеивает свет. Применяется для внутрипроизводственного контроля содержания сахара, основан на определении коэффициента преломления сахара, извлеченного из навески после удаления несахаров.
Реактивы и материалы. рефрактометр типа ИРФ-454, иммерсионная жидкость: альфа-хлор- или бромнафталин, вода дистиллированная, фильтры, пипетка, центрифуга.
Ход работы.
1. Твердые продукты. Откинуть осветительную призму. Очистить поверхность измерительной призмы и образца. На полированную поверхность образца нанести небольшую каплю иммерсионной жидкости и наложить его на измерительную призму. При наложении образца и умеренном нажиме на него иммерсионная жидкость должна распределяться равномерно по всей поверхности и не выступать за его края. Число интерференционных полос должно быть не более трех. Установка образца является идеальной при одноцветной окраске плоскости соприкосновения образца и призмы.
2.Густые продукты, у которых трудно отделить жидкую фазу, и темноокрашенные продукты следует разбавлять дистиллированной водой не более чем в два раза. При этом измельченную навеску густого продукта массой не менее 40 г разбавить водой, выдержать не менее 15 мин в кипящей водяной бане, затем смесь охладить, взвесить и отфильтровать как указано выше. Темноокрашенные жидкие продукты только перемешать с водой, определяя массу навески и смеси.
3.Жидкие продукты, не содержащие большого количества взвешенных частиц, используют для измерения. Жидкие продукты, содержащие большое количество взвешенных частиц, и пюреобразные продукты следует центрифугировать или фильтровать через несколько слоев марли, или слой ваты, или бумажный фильтр; первые порции фильтрата отбрасывать, а остальную часть необходимо использовать для измерений. Далее на чистую полированную поверхность измерительной призмы стеклянной палочкой или пипеткой осторожно, не касаясь призмы, нанести две-три капли жидкости. Опустить осветительную призму и прижать ее застежкой.
Измерения прозрачных жидкостей проводить в проходящем свете, когда он проходит через открытое окно осветительной призмы, при этом окно измерительной призмы закрыто зеркалом. Измерения окрашенных и мутных проб проводить в отраженном 'свете. Для этого закрыть заслонку и откинуть зеркало, с помощью которого направить свет в измерительную призму, при этом темное и светлое поля меняются местами. В остальном измерения следует проводить так же, как и для прозрачных жидкостей.
После установки исследуемого образца на измерительной призме навести окуляр на отчетливую видимость перекрестия. Вращением нижнего маховика границу светотени ввести в поле зрения окуляра. Вращать верхний маховик до исчезновения окраски граничной линии. Наблюдая в окуляр, нижним маховиком навести границу светотени точно на перекрестие и по шкале показателей преломления снять отсчет по неподвижному вертикальному штриху призмы. Измерения необходимо проводить при температуре 10-40°С, используя шкалу, градуированную в единицах массовой доли сахарозы. Во время измерений температуру следует поддерживать постоянной в пределах 0,5 ºС. При необходимости следует включить систему термосташрования призм рефрактометра и регулировать подачу воды так, чтобы выполнялись указанные выше условия. Температуру измеряемого раствора довести до значения, отличающегося от температуры призм не более чем на ±2°С. Необходимо проводить два параллельных измерения, принимая их среднее арифметическое значение.
Если продукт разбавлен водой, то массовую долю растворимых сухих веществ в продукте X следует вычислять по формуле:
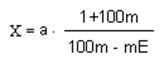
где а - значение массовой доли растворимых сухих веществ, полученное для разбавленного водой продукта, %; mi - масса добавленной воды, г; Е - массовая доля не растворимых в воде сухих веществ в продукте, %. Е ~ 5,5% - для томатной пасты с массовой долей растворимых сухих веществ 25-30%; Е = 5,0% - для сушеного винограда; Е = 1,8% - для джемов и повидла; Е = 0 - для темноокрашенных прозрачных жидких продуктов; m - масса навески продукта, г.
Если температура измерений отличается от 20 ºС, использовать поправку по справочной литературе.
Сахарометрический метод
Для определения концентрации экстрактивных веществ применяют ареометры сахарометры со шкалой 0-8, 8-16, 16-24% сухих веществ. Эти приборы представляют собой плавающий стеклянный цилиндрический сосуд, запаянный с обоих концов. Нижняя часть прибора заполнена свинцовой дробью, чтобы ареометр плавал строго вертикально. Верхняя часть ареометра - сахарометра представляет собой шкалу с делениями, градуированными по растворам чистой сахарозы при температуре 20 град С. Цена деления 0,1 % мас. В чистых растворах сахарозы сахарометры показывают процент растворенного сахара по массе ( г в 100г). В не чистых растворах (например в пивном сусле) они показывают видимое содержание сухих веществ в % мас. При отклонении температуры анализируемого раствора от 20 град С в показания сахарометра вносят поправку.
Оборудование и материалы: испытуемое сусло, асбестовая сетка, стеклянный цилиндр с поддоном, сахарометр.
Методика выполнения анализа: При анализе пробу сусла отбирают из сусловарочного аппарата перед перекачкой его на охлаждение, освобождают от дробины фильтрованием через сетку, охлаждают до 20 град С, и наливают в стеклянный цилиндр, диаметр которого больше диаметра сахарометра в 2-3 раза. Цилиндр ставят на поддон и плавно погружают сахарометр в сусло. Сахарометр должен быть предварительно очищен и высушен. При погружении сахарометра избыток сусла вытекает из цилиндра в поддон. Отсчет концетрации экстрактивных веществ по шкале сахарометра производят через 2-3 мин (необходимо для выравнивания температуры сусла и сахарометра) по верхнему мениску при положении глаза на уровне сусла в цилиндре.
Поляриметрический метод
Основан на измерении угла вращения плоскости поляризации луча света 1, прошедшего через оптически активную среду. В зависимости от направления вращения плоскости поляризации луча света бывают право- и левовращающие соединения и среды. Например, сахароза относится к правовращающим веществам ([a]D= + 66,50), а инвертный сахар — к левовращающим ([a]D= - 39,50).
В сахарной и крахмалопаточной промышленности наибольшее распространение получили специальные поляриметры-сахариметры. Пользуясь сахариметром, можно определить содержание сахарозы в сахаросодержащих продуктах в процентах.
Материалы и реактивы. Вода дистиллированная, навеска исследуемого продукта, фильтры, химический стакан, сахариметр.
Ход работы. Перед началом работы необходимо проверить нулевое положение прибора. Для этого вращают рукоятку кремальерной передачи и добиваются однородного поля зрения в обеих его половинах.
Взвешивают нормальную навеску продукта — 26,023 г — готовят раствор в мерной колбе вместимостью 100 см3, доводят дистиллированной водой температурой 20 °С до метки. В чисто вымытую и высушенную или сполоснутую исследуемым раствором трубку через воронку заливают исследуемый раствор температурой 20 °С так, чтобы верхний мениск его выступал над краями трубки. Выдерживают некоторое время, чтобы все содержащиеся в растворе пузырьки воздуха поднялись вверх. Подъем пузырьков можно ускорить, если слегка ударять пальцами по стенке трубки. Затем закрывают трубку покровным стеклом, надвигая его на торец трубки, как бы срезая раствор. Навинчивают гайку, следя за тем, чтобы под стеклом не остался воздушный пузырек. Тщательно протерев снаружи покровные стекла, помещают трубку в камеру прибора между поляризатором и анализатором. Устанавливают освещенность обеих половин поля зрения точно так же, как и при проверке нулевой точки. Затем производят отсчет показаний с необходимой точностью как по основной шкале, так и при помощи нониуса. Прежде чем зафиксировать результат, необходимо проверить, соответствует ли найденное положение компенсатора искомому: нужно едва заметным движением повернуть рукоятку сначала в одну сторону, а затем в другую. При этом происходит перемещение затемненной половины поля зрения с одной стороны на другую. Снова устанавливают одинаковую освещенность и фиксируют результат (отсчет производят не менее трех раз, каждый раз возвращая рукоятку в нулевое положение, и рассчитывают среднее арифметическое трех показаний). Устанавливают, какое деление нониуса точно совпадает с любым из делений основной шкалы.
Это будет α.
Известно,
что оптическая активность веществ характеризуется углом удельного вращения ![]() (D
желтая линия натрия с длиной волны 588 нм) – устойчивой величиной угла
вращения плоскости поляризации света (зависящей от длины волны света и
температуры) раствором вещества при его концентрации С, которая
равняется 1 г вещества в 1 мл раствора, при толщине слоя l
дм [для данного опыта искомое есть С, значение которого затем возводят в
проценты]:
(D
желтая линия натрия с длиной волны 588 нм) – устойчивой величиной угла
вращения плоскости поляризации света (зависящей от длины волны света и
температуры) раствором вещества при его концентрации С, которая
равняется 1 г вещества в 1 мл раствора, при толщине слоя l
дм [для данного опыта искомое есть С, значение которого затем возводят в
проценты]:
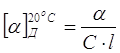
Градусы линейной шкалы сахариметра можно перевести в градусы круговой шкалы поляриметра при помощи следующих соотношений: градус круговой шкалы поляриметра соответствует 2,883° линейной шкалы сахариметра. Либо наоборот: градус линейной шкалы сахариметра равен 0,3468° круговой шкалы поляриметра.
Приборы и устройства для физико-химического определения сахаров прямо на поточных линиях
За последние годы получило развитие специальное приборостроение для пищевой промышленности. В связи с тем, что среды пищевой промышленности, а также производственные помещения, оборудование, технологические процессы обладают определенными специфическими свойствами взрывоопасностью, вязкостью, налипанием, абразивностью и т. п. — необходимо, чтобы применяемые приборы и средства автоматизации своими характеристиками удовлетворяли перечисленным условиям.
Автоматические рефрактометры широко применяются в различных отраслях пищевой промышленности (сахарной, спиртовой, консервной, кондитерской, винодельческой) — как и обыкновенные — для определения концентрации растворенных в жидкостях веществ. Принцип действия этих приборов основан на использовании зависимости показателя преломления бинарной смеси от соотношения ее компонентов. Существует несколько методов определения показателя преломления, из которых наиболее приемлемым считается спектрометрический, основанный на использовании полного внутреннего отражения. Рефрактометры полного внутреннего отражения могут применяться для работы с непрозрачными жидкостями, что является очень важным при контроле многих технологических сред пищевых производств. Ниже показана принципиальная схема рефрактометра типа РДА:
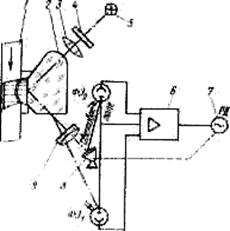
Он основан на использовании полного внутреннего отражения. В трубопроводе 1, по которому протекает анализируемая жидкость, установлена измерительная призма 2, на которую поступает поток света от источника 5, проходящий предварительно светофильтр 4 и коллиматор 3. Световой поток, попадая на границу раздела среды и призмы, отражается от нее и идет в направлении оптического рассеивателя 9, пройдя который, поступает на фотоэлемент ФЭ, и зеркало 8. Поток, отраженный от зеркала, попадает на фотоэлемент ФЭ2. Сигнал разбаланса, равный разности ЭДС от фотоэлементов, усиливается электронным усилителем 6 и поступает на реверсивный двигатель 7, с осью которого связано отсчетное устройство, не показанное по схеме. Двигатель 7 поворачивает зеркало 8 до тех пор, пока свет, направляемый на фотоэлемент ФЭ2, не уравновесит световой поток, падающий на ФЭ и тем самым не приведет систему в равновесие.
Основная погрешность измерения ±0,5-1,5 % сухих веществ.
Автоматические поляриметры. Ниже показана схема автоматического поляриметра для непрерывного анализа пищевых сред:
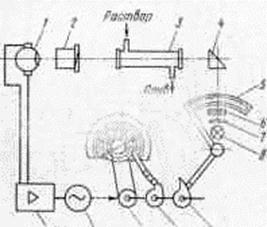
Луч света от источника 8 проходит через конденсор 7 и светофильтр 6 и попадает на поляризатор 5, откуда выходит плоскополяризованным. Затем поток поляризованного света с помощью призмы 4 направляется в кювету 3, через которую непрерывно протекает контролируемый раствор. После кюветы свет проходит поляроидный анализатор и магнитооптический модулятор 2 и направляется на фотоприемник 1, включенный на вход электронного усилителя 13. Если оптически активные вещества отсутствуют в анализируемой жидкости, свет полностью гасится на анализаторе 2, не попадая на фотоэлемент. Появление оптически активного вещества в растворе вызывает поворот плоскости поляризации на угол, пропорциональный количеству этого вещества, и модулированный поток света падает на фотоэлемент, благодаря чему на входе в усилитель 13 возникает сигнал разбаланса, который усиливается и приводит во вращение реверсивный двигатель 12, перемещающий через кулачок 9 поляризатор 5. Вращение происходит до тех пор, пока не будет скомпенсирован возникший поворот плоскости поляризации. Следовательно, угол поворота поляризатора 5 прямо пропорционален содержанию в анализируемом растворе оптически активного углевода. Насаженные на ось реверсивного двигателя кулачок 10 и ролик 11 предназначены для передачи движения на показывающие устройства.
По приведенной схеме работают приборы для определения содержания сахара в сахарной свекле, продуктах, полупродуктах и отходах сахарного производства (тип САП), имеющие пределы измерения 7-20 °S по международной сахарной шкале и предельно допустимую погрешность ±1°S. Специальные печатающие устройства этих приборов отбивают содержание сахаристых веществ на чеке, вручаемом сдатчику сахарной свеклы на свеклоприемных пунктах.
Разработаны и применяются поляриметры для определения кристалличности ирисных масс и некоторых других материалов.
Основная погрешность измерения ±0,9-1,7 % сухих веществ.
Анализатор автоматический колориметрический устройство контроля качественных показателей полупродуктов и готовой продукции в процессе производства. Это анализатор сбраживаемых углеводов периодического действия, коротко зовущийся АГК (анализатор гексоз колориметрический), предназначен для автоматического циклического измерения концентрации растворимых сбраживаемых углеводов (гексоз) в фильтратах зрелых зерно-картофельных бражек в бродильных отделениях спиртовых заводов, перерабатывающих крахмалистое сырье (зерно, картофель). АГК может быть применен для оперативного или автоматического контроля хода брожения по содержанию сбраживаемых углеводов в бражке при автоматизации технологических процессов. Может быть также применен в качестве экспресс-анализатора при приготовлении проб фильтрата бражки вручную (фильтрованием) или как датчик автоматического контроля содержания сбраживаемых углеводов в зрелых бражках бродильных батарей при совместной работе с автоматическим фильтром-пробоотборником бражки типа ФПА, осуществляющим циклически в заданном темпе автоматический отбор пробы бражки из бродильной батареи, ее фильтрацию и подачу в анализатор.
В основу принципа действия АГК положен антроновый метод.
Анализатор по конструктивному исполнению представляет собой единую систему, включающую в себя ряд функциональных узлов (блоков), связанных общей функциональной схемой. Он устанавливается в бродильном отделении спирт-завода у щита автоматики или в другом хорошо освещенном месте. Диапазон измерения концентрации растворимых сбраживаемых углеводов от 0,10 до 0,60 г на 100 мл. Класс точности прибора 10. Время установления показаний в каждом цикле измерения (без учета времени приготовления пробы реакционной смеси) не более 180 с. Выходной сигнал аналоговый постоянного тока 0-5 мА. Длительность цикла не более 20 мин.
Основным достоинством АГК является возможность измерения концентрации сбраживаемых углеводов (гексоз) в фильтратах бражек в присутствии сходных с ними по физико-химическим свойствам несбраживаемых углеводов с исключением влияния последних на результат определения сбраживаемых углеводов.
Плотномеры. Единицей измерения плотности в Международной системе (СИ) является килограмм на кубический метр (кг/м3). Измерение плотности жидкости имеет большое значение в автоматизации целого ряда процессов сахарного, масло-жирового, кондитерского, спиртового, винодельческого, пивоваренного и других производств, требующих непрерывного контроля плотности. Приборы для измерения плотности жидкостей, растворов и пульп называются плотномерами.
Принцип действия радиоизотопных плотномеров основан на определении изменения интенсивности потока ץ-лучей после прохождения их через измеряемую среду. В качестве примера радиоизотопного плотномера может служить прибор типа ПР- 1024В, предназначенный для измерения плотности в потоке жидкостей, растворов, суспензий:
Плотномер состоит из блока источника излучения 1, блока приемника 3, электронного самопишущего блока 4 и стабилизатора напряжения.
Блок источника излучения представляет собой чугунный корпус, залитый свинцом и являющийся надежной защитой обслуживающего персонала от ионизирующего излучения. Внутри корпуса помещается источник ионизирующего излучения (ампула с изотопом цезия-137: период полураспада 1,5 года), который перемещают в нужное положение специальным механизмом.
Блок приемника в виде стальной коробчатой конструкции состоит из основания и корпуса, служащего для защиты от механических повреждений. На основании крепится плита с узлами и деталями приемника. Электронный самопишущий блок выполнен на базе электронного моста КСМ-3. При помощи разъемов типа ШР осуществляется электрическое соединение электронного самопишущего блока с остальными блоками прибора. Контейнер основного источника и блок приемника крепятся на общем контейнере, обеспечивающем их фиксированное положение.
Плотномер работает следующим образом: поток γ-лучей от источника излучения 1, пройдя через контролируемую среду 2, регистрируется блоком приемника 3. В блоке приемника ץ-лучи преобразуются в электрические импульсы, которые в электронном самопишущем блоке усиливаются, формируются и подаются на вход электронного моста, шкала которого отградуирована по системе СИ.
Пределы измерения плотности 500-3000 кг/м3. Основная погрешность прибора не более ±2 %. Внутренний диаметр трубопровода, на котором может быть установлен плотномер, 100-300 мм.
© 2009 База Рефератов