
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Курсовая работа: Электролиты и их свойства
Курсовая работа: Электролиты и их свойства
Введение
Водные растворы солей, кислот и оснований обладают некой особенностью — они проводят электрический ток. При этом безводные твердые соли и основания, а также безводные кислоты тока не проводят; почти не проводит тока и чистая вода. Очевидно, что при растворении в воде подобные вещества подвергаются каким-то глубоким изменениям, которые и обусловливают электропроводность получаемых растворов.
Например, при прохождении тока через раствор серной кислоты, происходит разложение воды на составные части – водород и кислород, выделяющиеся на пластинах, соединенных соответственно с отрицательным и положительным полюсами батареи. Такого рода растворы, разлагающиеся химически при прохождении через них тока, будем называть электролитами, а сам процесс разложения вещества электрическим током электролизом.
1. Определение электролитов
Можно сказать, что электролиты – это вещества, в которых в заметной концентрации присутствуют ионы, обусловливающие прохождение электрического тока (ионную проводимость). Электролиты также имеют название проводников второго рода.
В узком смысле слова электролиты – вещества, молекулы которых в растворе, вследствие электролитической диссоциации, распадаются на ионы. Среди электролитов различают твердые, растворы электролитов и ионные расплавы. Растворы электролитов часто также называют электролиты. В зависимости от вида растворителя электролиты делятся на водные и электролиты неводные. К особому классу относятся высокомолекулярные электролиты – полиэлектролиты.
В соответствии с природой ионов, образующихся при электролитической диссоциации водных растворов, выделяют солевые электролиты (в них отсутствуют ионы Н+ и ОН-), кислоты (преобладают ионы Н+) и основания (преобладают ионы ОН-). Если при диссоциации молекул электролитов число катионов совпадает с числом анионов, то такие электролиты называют симметричными (1,1 -валентными, например, КСl, 2,2-валентными, например, CaSO4, и т.д.). В противном случае электролиты называют несимметричными (1,2-валентные электролиты, напр. H2SO4, 3,1-валентные, например, А1(ОН)3, и т.д.). В зависимости от способности к электролитической диссоциации электролиты условно разделяют на сильные и слабые. Слабые электролиты характеризуются, прежде всего, константой и степенью диссоциации, а сильные активностью ионов.
1.1 Слабые электролиты. Константа и степень диссоциации
Молекулы слабых электролитов лишь частично диссоциированы на ионы, которые находятся в динамическом равновесии с недиссоциирующими молекулами. К слабым электролитам относятся многие органические кислоты и основания в водных и неводных растворителях. Степень диссоциации зависит от природы растворителя, концентрации раствора, температуры и других факторов( <1). Один и тот же электролит при одинаковой концентрации, но в различных растворителях образует растворы с различной степенью диссоциации.
В растворах слабых электролитов устанавливается равновесие между недиссоциированными молекулами и продуктами их диссоциации — ионами. Например, в водном растворе уксусной кислоты устанавливается равновесие
![]()
![]() СНзСООН Н+ + СН3СОО-
СНзСООН Н+ + СН3СОО-
Константа которого (константа диссоциации) связана с концентрациями соответствующих частиц соотношением:
К = [н+] [сн3соо-]
[сн3соо]
Степенью диссоциации а электролита называется доля его молекул, подвергшихся диссоциации, т. е. отношение числа молекул, распавшихся в данном растворе на ионы, к общему числу молекул электролита в растворе.
В случае электролита MX, диссоциирующего на ионы М+ и Х-, константа и степень диссоциации связаны соотношением (закон разбавления Оствальда)
К = а2С/1 - а
где С — молярная концентрация электролита, моль/л.
Если степень диссоциации значительно меньше единицы, то при
приближенных вычислениях можно принять, что 1 - а![]() 1. Тогда выражение закона
разбавления упрощается:
1. Тогда выражение закона
разбавления упрощается:
К = а2С
Отсюда
![]()
![]()
Последнее соотношение показывает, что при разбавлении раствора (т. е. при уменьшении концентрации электролита С) степень диссоциации электролита возрастает.
Если в растворе электролита MX степень его диссоциации равна а, то концентрации ионов М+ и Х- в растворе одинаковы и составляют:
[М+]=[Х-]=аС
Подставив сюда значение а из предыдущего соотношения, находим:
[М+]=[Х-]=С![]() =
=![]()
Для расчетов, связанных с диссоциацией кислот, часто удобно пользоваться не константой К, а так называемым показателем константы диссоциации рК, который определяется соотношением
рК = - lg К
Очевидно, что с возрастанием К, т. е. с увеличением силы кислоты, значение рК уменьшается; следовательно, чем больше рК, тем слабее кислота.
При введении в раствор слабого электролита одноименных ионов (т. е. ионов, одинаковых с одним из ионов, образующихся при диссоциации электролита) равновесие диссоциации нарушается и смещается в направлении образования недиссоциированных молекул, так что степень диссоциации электролита уменьшается. Так, прибавление к раствору уксусной кислоты ее соли (например, ацетата натрия) приведет к повышению концентрации ионов СН3СОО - и, в соответствии с принципом Ле Шателье, равновесие диссоциации сместится влево.
![]()
![]() СНзСООН Н+ + СН3СОО-
СНзСООН Н+ + СН3СОО-
В растворах многоосновных кислот, а также оснований, содержащих несколько гидроксильных групп, устанавливаются ступенчатые равновесия, отвечающие последовательным стадиям диссоциации. Так, диссоциация ортофосфорной кислоты протекает в три ступени, каждой из которых отвечает определенное значение ступенчатой константы диссоциации. Поскольку К1 >> К2 >>К3, то в наибольшей степени протекает диссоциация по первой ступени, а при переходе к каждой последующей стадии степень диссоциации, как правило, резко уменьшается.
Н3РО4 ↔Н+ + Н2РО4- (К1=7,5 . 10-3)
Н2РО4- ↔ Н+ + НРО42- (К2=6,3 . 10-8)
НРО42- ↔ Н+ + РО43- (К3=1,3 . 10-12)
Диссоциация электролита приводит к тому, что общее число частиц растворенного вещества (молекул и ионов) в растворе возрастает по сравнению с раствором неэлектролита той же молярной концентрации. Поэтому свойства, зависящие от общего числа находящихся в растворе частиц растворенного вещества (коллигативные свойства), — такие, как осмотическое давление, понижение давления пара, повышение температуры кипения, понижение температуры замерзания — проявляются в растворах электролитов в большей степени, чем в равных по концентрации растворах неэлектролитов. Если в результате диссоциации общее число частиц в растворе электролита возросло в i раз по сравнению с числом его молекул, то это должно быть учтено при расчете осмотического давления и других коллигативных свойств. Формула для вычисления понижения давления ∆р пара растворителя приобретает в этом случае следующий вид:
∆р= р0 in2/п1+ in2
Здесь
р0 — давление насыщенного пара над чистым растворителем;
п2 — число молей растворенного вещества;
п1 — число молей растворителя;
i — изотонический коэффициент или коэффициент Вант-Гоффа.
Аналогично понижение температуры кристаллизации ∆tкрист и повышение температуры кипения ∆tкип раствора электролита находят по формулам
∆tкрист =iKm
∆tкип =iEm
где m — моляльная концентрация электролита, а К и Е — соответственно, криоскопическая постоянная и эбуллиоскопическая постоянная растворителя.
Наконец, для вычисления осмотического давления (Р, кПа) раствора электролита используют формулу
Р =iCRT
где С — молярная концентрация электролита, моль/л; R — газовая постоянная (8,31 Дж . моль-1 . К-1); Т — абсолютная температура, К.
Нетрудно видеть, что изотонический коэффициент i может быть вычислен как отношение ∆р , ∆tкрист, ∆tкип, Р, найденных на опыте, к тем же величинам, вычисленным без учета диссоциации электролита (∆рвыч, ∆tкрист.выч, ∆tкип.выч, Рвыч):
i=∆р/∆рвыч=∆tкрист/∆tкрист.выч=∆tкип/∆tкип.выч=Р/ Рвыч
Изотонический коэффициент i связан со степенью диссоциации электролита а соотношением
i=1+a (k - 1) или ![]()
где k — число ионов, на которые распадается при диссоциации молекула электролита (для КСl k = 2, для ВаСl2 и Na2SO4 k =3 и т. д.).
Таким образом, найдя по опытным величинам ∆р, ∆tкрист и т. п. значение i, можно вычислить степень диссоциации электролита в данном растворе. При этом следует иметь в виду, что в случае сильных электролитов найденное таким способом значение а выражает лишь «кажущуюся» степень диссоциации, поскольку в растворах сильные электролиты диссоциированы полностью. Наблюдаемое отличие кажущейся степени диссоциации от единицы связано с межионными взаимодействиями в растворе.
1.2 Сильные электролиты. Активность ионов
Сильными электролитами называются такие электролиты, степень диссоциации которых в растворах равна единице (т.е. диссоциируют полностью) и не зависит от концентрации раствора. К ним относятся большинство солей, которые уже в кристаллическом состоянии построены из ионов, гидроксиды щелочных и щелочноземельных металлов, некоторые кислоты (НСl, HBr, HI, HClO4, HNO3).
В растворах сильных электролитов концентрация ионов довольно велика, так что силы межионного взаимодействия заметно проявляются даже при малой концентрации электролита. В результате ионы оказываются не вполне свободными в своем движении, и все свойства электролита, зависящие от числа ионов, проявляются слабее, чем следовало бы ожидать при полной диссоциации электролита на не взаимодействующие между собой ионы. Поэтому для описания состояния ионов в растворе пользуются, наряду с концентрацией ионов, их активностью, т. е. условной (эффективной) концентрацией ионов, в соответствии с которой они действуют в химических процессах. Активность иона а (моль/л) связана с его молярной концентрацией в растворе С соотношением
a = fC
где f — коэффициент активности иона (безразмерная величина).
![]() Коэффициенты активности ионов зависят от состава и концентрации
раствора, от заряда и природы лона и от других условий. Однако в разбавленных
растворах (С ≤ 0,5 моль/л) природа иона слабо сказывается на величине его
коэффициента активности. Приближенно можно считать, что в разбавленных
растворах коэффициент активности иона в данном растворителе зависит только от
заряда иона и ионной силы раствора I, которая равна полусумме произведений концентрации С каждого иона на
квадрат его заряда z:
Коэффициенты активности ионов зависят от состава и концентрации
раствора, от заряда и природы лона и от других условий. Однако в разбавленных
растворах (С ≤ 0,5 моль/л) природа иона слабо сказывается на величине его
коэффициента активности. Приближенно можно считать, что в разбавленных
растворах коэффициент активности иона в данном растворителе зависит только от
заряда иона и ионной силы раствора I, которая равна полусумме произведений концентрации С каждого иона на
квадрат его заряда z:
I=0,5 (С1 z12 + С2 z22 + …+ Сn zn2)=0,5 Сi z i2
В табл. 1 приведены значения
коэффициентов активности ионов в разбавленных растворах в зависимости от их
заряда и ионной силы раствора. Приближенно коэффициент активности иона в
разбавленном растворе можно также вычислить по формуле: lg f = — 0,5z2 - ![]() .
.
Таблица 1.
| Ионная сила раствора I | Заряд иона z | ||
| ±1 | ±2 | ±3 | |
|
0,001 0,002 0,005 0,01 0,02 0,05 0,1 0,2 0,3 |
0,98 0,97 0,95 0,92 0,90 0,84 0,81 0,80 0,81 |
0,78 0,74 0,66 0,60 0,53 0,50 0,44 0,41 0,42 |
0,73 0,66 0,55 0,47 0,37 0,21 0,16 0,14 0,14 |
2. Свойства электролитов
Электролиты по своей структуре представляют собой сложные системы, состоящие из ионов, окруженных молекулами растворителя, недиссоциированных молекул растворенного вещества, ионных пар и более крупных агрегатов. Свойства электролитов определяются характером ион-ионных и ион-молекулярных взаимодействий, а также изменением свойств и структуры растворителя под влиянием растворенных частиц электролитов.
В зависимости от концентрации
электролитов выделяют область разбавленных растворов, которые по своей
структуре близки к структуре чистого растворителя, нарушаемой, однако,
присутствием и влиянием ионов; переходную область и область концентрированных
растворов. Весьма разбавленные растворы слабых электролитов по своим свойствам
близки к идеальным растворам и достаточно хорошо описываются классической
теорией электролитической диссоциации. Разбавленные растворы сильных
электролитов заметно отклоняются от свойств идеальных растворов, что
обусловлено электростатическим межионным взаимодействие. Их описание проводится
в рамках Дебая-Хюккеля теории, которая удовлетворительно объясняет
концентрационную зависимость термодинамических свойств - коэффициент активности
ионов, осмотический коэффициент и др., а также неравновесных свойств - электропроводности,
диффузии, вязкости. ![]()
Изучение свойств электролитов важно для выяснения механизмов электролиза, электрокатализа, электрокристаллизации, коррозии металлов и др., для совершенствования механизмов разделения веществ - экстракции и ионного обмена. Исследование свойств электролитов стимулируется энергетическими проблемами (создание новых топливных элементов, солнечных батарей, электрохимических преобразователей информации), а также проблемами защиты окружающей среды.
2.1 Ионная проводимость электролитов
Факт разложения электролитов при прохождении через них тока показывает, что в них движение зарядов сопровождается движением атомов или групп атомов, связанных друг с другом (например, SO4, NO3 и т. п.); эти атомы или атомные группы представляют собой части молекулы растворенного вещества.
Естественно предположить, что заряжены именно эти части молекулы в растворе и что они являются носителями электрического заряда. Их перемещение под действием сил электрического поля и представляет собой электрический ток, идущий через электролит.
Было обнаружено, что при прохождении тока через электролит выделение вещества происходит на обоих электродах. По химическому составу это разные части молекулы растворенного вещества. По количеству, если измерять его в химических эквивалентах, они равны. Знаки зарядов у них, очевидно, противоположны.
Мы знаем, что заряженные атомы называются ионами. То же название носят заряженные молекулы или их части. Мы можем, следовательно, сказать, что проводимость электролитов является ионной, т. е. обусловлена движением в них положительных и отрицательных ионов, которые образуются из нейтральной молекулы путем распада ее на две части, заряженные равными и противоположными зарядами.
Молекулы растворенного вещества, которые до растворения были электрически нейтральны, при растворении распадаются на положительные и отрицательные ионы, способные перемещаться независимо друг от друга.
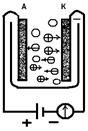
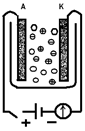
а) б)
Рис. 1
Проводимость электролита зависит от наличия положительных и отрицательных ионов (кружки со знаками «+» или «-»): а) цепь разомкнута, тока нет; б) цепь замкнута, через электролит идет ионный ток.
Эти представления иллюстрируются рис. 1. Кружками между электродами со значками «+» и «-» схематически изображены положительные и отрицательные ионы растворенного вещества. Пока между электродами А и К не создано поле, ионы эти совершают только беспорядочное тепловое движение, как и все остальные молекулы раствора (рис.1, а). В каждом направлении за единицу времени протекает одинаковый положительный и отрицательный заряд, т. е. нет электрического тока — преимущественного переноса заряда в определенном направлении. При наложении разности потенциалов на электроды А и К, когда внутри электролита возникает электрическое поле, на это беспорядочное движение накладывается упорядоченное движение в противоположные стороны ионов различных знаков: отрицательных — к аноду А, положительных — к катоду К (рис. 1, б).
При соприкосновении с катодом положительные ионы получают недостающие им электроны и выделяются в виде нейтральных атомов, а взамен электронов, нейтрализовавших ионы, новые электроны переходят от батареи к катоду. Точно так же отрицательные ионы при соприкосновении с анодом отдают ему свои избыточные электроны, превращаясь в нейтральные атомы; электроны же уходят по металлическим проводам в батарею. Таким образом, ток в электролите обусловлен движущимися ионами; на электродах же происходит нейтрализация ионов и выделение их в виде нейтральных атомов (или молекул). Итак, электрический ток в электролитах представляет собой движение положительных и отрицательных ионов.
Такое представление об электролизе подкрепляется многочисленными фактами. С этой точки зрения первый закон Фарадея получает простое объяснение. Каждый осаждающийся на электроде ион переносит с собой некоторый электрический заряд. Это значит, что полный заряд, перенесенный всеми ионами, должен быть пропорционален полному числу ионов, осевших на электродах, т. е. массе выделенного вещества. А это и есть первый закон Фарадея. Так же естественно и просто объясняется с этой точки зрения и второй закон Фарадея, дающий возможность вычислить электрический заряд, связанный с каждым ионом.
Отметим, что название «ион» введено Фарадеем (от греческого слова «ион» — идущий). Ионы, заряженные положительно и выделяющиеся на катоде, Фарадей назвал катионами, ионы, выделяющиеся на аноде,— анионами.
Опыт показал, что водород и металлы всегда выделяются на катоде; это значит, что в электролитах водород и металлы образуют положительные, ионы.
2.2 Движение ионов в электролитах
Движение ионов в электролитах в некоторых случаях может быть показано весьма наглядно.
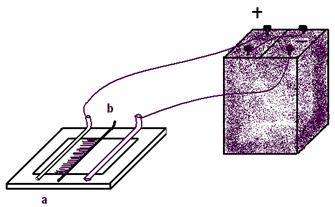
Рис. 2. Опыт, показывающий движение ионов. Листок фильтровальной бумаги пропитан раствором электролита и фенолфталеина, ab — нитка, смоченная раствором электролита
Пропитаем листок фильтровальной бумаги раствором электролита (сернокислого натра, Na2SO4) и фенолфталеина и поместим на стеклянную пластинку (рис. 2).
Поперек бумаги положим обыкновенную белую нитку, смоченную раствором едкого натра (NaOH). Бумага под ниткой окрасится в малиновый цвет благодаря взаимодействию ионов гидроксила (ОН) из NaOH с фенолфталеином. Затем прижмем к краям листка проволочные электроды, присоединенные к гальваническому элементу, и включим ток.
Ионы гидроксила из едкого натра начнут двигаться к аноду, окрашивая бумагу в малиновый цвет. По скорости перемещения малинового края можно судить о средней скорости движения ионов под влиянием электрического поля внутри электролита. Опыт показывает, что эта скорость пропорциональна напряженности поля внутри электролита. При заданном поле эта скорость для разных ионов несколько различна. Но, в общем, она невелика и для обычно применяющихся полей измеряется сотыми и даже тысячными долями сантиметра в секунду.
2.3 Теория электролитической диссоциации
Сванте Аррениус обратил внимание на тесную связь между
способностью растворов солей, кислот и оснований проводить электрический ток и
отклонениями растворов этих веществ от законов Вант-Гоффа и Рауля. Он показал, что
по электропроводности раствора можно рассчитать величину его осмотического
давления, а, следовательно, и поправочный коэффициент i. Значения
i, вычисленные им из электропроводности, хорошо совпали с
величинами, найденными для тех же растворов иными методами.![]()
Причиной чрезмерно высокого осмотического давления растворов электролитов является, согласно Аррениусу, диссоциация электролитов на ионы. Вследствие этого, с одной стороны, увеличивается общее число частиц в растворе, а, следовательно, возрастают осмотическое давление, понижение давления пара и изменения температур кипения и замерзания, с другой, — ионы обусловливают способность раствора проводить электрический ток.
Эти предположения в дальнейшем были развиты в стройную теорию, получившую название теории электролитической диссоциации. Согласно этой теории, при растворении в воде электролиты распадаются (диссоциируют) на положительно и отрицательно заряженные ионы. Положительно заряженные ионы называются катионами; к ним относятся, например, ионы водорода и металлов. Отрицательно заряженные ионы называются анионами; к ним принадлежат ионы кислотных остатков и гидроксид-ионы. Как и молекулы растворителя, ионы в растворе находятся в состоянии неупорядоченного теплового движения.
Процесс электролитической диссоциации изображают, пользуясь химическими уравнениями. Например, диссоциация НСl выразится уравнением:
НСl = Н+ + Сl-
Распад электролитов на ионы объясняет отклонения от законов Вант-Гоффа и Рауля. В качестве примера можно привести понижение температуры замерзания раствора NaCl. Теперь нетрудно понять, почему понижение температуры замерзания этого раствора столь велико. Хлорид натрия переходит в раствор в виде ионов Na+ и Сl-. При этом из одного моля NaCl получается не 6,02 • 1023 частиц, а вдвое большее их число. Поэтому и понижение температуры замерзания в растворе NaCl должно быть вдвое больше, чем в растворе неэлектролита той же концентрации.
Точно так же в очень разбавленном растворе хлорида бария, диссоциирующего согласно уравнению осмотическое давление оказывается в 3 раза больше, чем вычисленное по закону Вант-Гоффа, так как число частиц в растворе в 3 раза больше, чем, если бы хлорид бария находился в нем в виде молекул ВаСl2.
ВаСl2=Ва2+ + 2Сl-
Таким образом, особенности водных растворов электролитов, противоречащие с первого взгляда законам Вант-Гоффа и Рауля, были объяснены на основе этих же законов.
Однако теория Аррениуса не учитывала всей сложности явлений в растворах. В частности, она рассматривала ионы как свободные, независимые от молекул растворителя частицы. Теории Аррениуса противостояла химическая, или гидратная, теория растворов Менделеева, в основе которой лежало представление о взаимодействии растворенного вещества с растворителем. В преодолении кажущегося противоречия обеих теорий большая заслуга принадлежит русскому ученому И. А. Каблукову, впервые высказавшему предположение о гидратации ионов. Развитие этой идеи привело в дальнейшем к объединению теорий Аррениуса и Менделеева.
2.4 Процесс диссоциации
В зависимости от структуры растворяющегося вещества в безводном состоянии его диссоциация протекает по-разному. Наиболее типичны при этом два случая. Один из них это диссоциация растворяющихся солей, т. е. кристаллов с ионной структурой, второй—диссоциация при растворении кислот, т. е. веществ, состоящих из полярных молекул.
Когда кристалл соли, например, хлорида калия, попадает и воду, то расположенные на его поверхности ионы притягивают к себе полярные молекулы воды (ион-дипольное взаимодействие). К ионам калия молекулы воды притягиваются своими отрицательными полюсами, а к хлорид-ионам- положительными. Но, если ионы притягивают к себе молекулы воды, то и молекулы воды с такой же силой притягивают к себе ионы. В то же время притянутые молекулы поды испытывают толчки со стороны других молекул, находящихся в движении. Этих толчков вместе с тепловыми колебаниями ионов в кристалле оказывается достаточно для отделения ионов от кристалла и: перехода их в раствор. Вслед за первым слоем ионов в раствор переходит следующий слой, и таким образом идет постепенное растворение кристалла.
Иначе протекает диссоциация полярных молекул. Молекулы воды, притянувшиеся к концам полярной молекулы (диполь-дипольное взаимодействие), вызывают расхождение ее полюсов поляризуют молекулу. Такая поляризация в сочетании с колебательным тепловым движением атомов в рассматриваемой молекуле, а также с непрерывным тепловым движением окружающих ее молекул воды приводит, в конечном счете, к распаду полярной молекулы на ионы. Как и в случае растворения ионного кристалла, эти ионы гидратируются. При этом ион водорода Н+ (т. е. протон) оказывается прочно связанным с молекулой воды в ион гидроксония Н3О+. Так, при растворении в воде хлороводорода происходит процесс, который схематически можно выразить уравнением
Н2О + НСl = Н3О+ +Сl-
В результате этого процесса молекула НСl расщепляется таким образом, что общая пара электронов остается у атома хлора, который превращается в ион Сl-, а протон, внедряясь в электронную оболочку атома кислорода в молекуле воды, образует ион гидроксония Н3О+.
Подобного же рода процессы происходят и при растворении в воде других кислот, например, азотной:
Н2О + НNO3 = Н3О+ + NO3-
Перешедшие в раствор ионы остаются связанными с молекулами воды и образуют гидраты ионов. Иначе говоря, в результате диссоциации образуются не свободные ионы, а соединения ионов с молекулами растворителя. В общем случае любого растворителя эти соединения называются сольватами ионов. Но в уравнениях диссоциации обычно пишут формулы ионов, а не их гидратов или сольватов, тем более что число молекул растворителя, связанных с ионами, изменяется в зависимости от концентрации раствора и других условий.
Диссоциации веществ как ионного, так и молекулярного строения способствует полярность молекул растворителя. Поэтому не только вода, но и другие жидкости, состоящие из полярных молекул (муравьиная кислота, этиловый спирт, аммиак и другие), также являются ионизирующими растворителями: соли, кислоты и основания, растворенные в этих жидкостях, диссоциируют на ионы.
2.5 Свойства кислот, оснований и солей с точки зрения теории электролитической диссоциации
Рассмотрим в свете теории электролитической диссоциации свойства веществ, которые в водных растворах проявляют свойства электролитов.
Кислоты. Для кислот характерны следующие общие свойства:
Ø способность взаимодействовать с основаниями с образованием солей;
Ø способность взаимодействовать с некоторыми металлами с выделением водорода;
Ø способность изменять цвета индикаторов, в частности, вызывать красную окраску лакмуса;
Ø кислый вкус.
При диссоциации любой кислоты образуются ионы водорода. Поэтому все свойства, которые являются общими для водных растворов кислот, мы должны объяснить присутствием гидратированных ионов водорода. Это они вызывают красный цвет лакмуса, сообщают кислотам кислый вкус и т. д. С устранением ионов водорода, например при нейтрализации, исчезают и кислотные свойства. Поэтому теория электролитической диссоциации определяет кислоты как электролиты, диссоциирующие в растворах с образованием ионов водорода.
У сильных кислот, диссоциирующих нацело, свойства кислот проявляются в большей степени, у слабых — в меньшей. Чем лучше кислота диссоциирует, т. е. чем больше ее константа диссоциации, тем она сильнее.
Величины констант диссоциации кислот изменяются в очень широких пределах. В частности, константа диссоциации циановодорода много меньше, чем уксусной кислоты. И хотя обе эти кислоты — слабые, все же уксусная кислота значительно сильнее циановодорода. Величины первой и второй констант диссоциации серной кислоты показывают, что в отношении первой ступени диссоциации H2SO4 — сильная кислота, а в отношении второй — слабая. Кислоты, константы диссоциации которых лежат в интервале 10-4– 10-2, иногда называют кислотами средней силы. К ним, в частности, относятся ортофосфорная и сернистая кислоты (в отношении диссоциации по первой ступени).
Основания. Водные растворы оснований обладают следующими общими свойствами:
Ø способностью взаимодействовать с кислотами с образованием солей;
Ø способностью изменять цвета индикаторов иначе, чем их изменяют кислоты (например, они вызывают синюю окраску лакмуса);
Ø Своеобразным «мыльным» вкусом.
Поскольку общим для всех растворов оснований является присутствие в них гидроксид-ионов, то ясно, что носителем основных свойств является гидроксид-ион. Поэтому с точки зрения теории электролитической диссоциации основания— это электролиты, диссоциирующие в растворах с отщеплением гидроксид-ионов.
Сила оснований, как и сила кислот, зависит от величины константы диссоциации. Чем больше константа диссоциации данного основания, тем оно сильнее.
Существуют гидроксиды, способные вступать во взаимодействие и образовывать соли не только с кислотами, но и с основаниями. К таким гидроксидам принадлежит гидроксид цинка. При взаимодействии его, например, с соляной кислотой получается хлорид цинка:
Zn(OH)2 + 2НСl = ZnСl2 + 2Н2О
а при взаимодействии с гидроксидом натрия — цинкат натрия:
Zn(OH)2 + 2NaOH = Na2ZnО2+ 2Н2О
Гидроксиды, обладающие этим свойством, называются амфотерными гидроксидами или амфотерными электролитами. К таким гидроксидам, кроме гидроксида цинка, относятся гидроксиды алюминия, хрома и некоторые другие.
Явление амфотерности объясняется тем, что в молекулах амфотерных электролитов прочность связи между металлом и кислородом незначительно отличается от прочности связи между кислородом и водородом. Диссоциация таких молекул возможна, следовательно, по местам обеих этих связей. Если обозначить амфотерный электролит формулой ROH, то его диссоциацию можно выразить схемой
Н+ + RO- ↔ ROH↔R+ + OH-
Таким образом, в растворе амфотерного электролита существует сложное равновесие, в котором участвуют продукты диссоциации как по типу кислоты, так и по типу основания.
Явление амфотерности наблюдается также среди некоторых органических соединений. Важную роль оно играет в биологической химии; например, белки — амфотерные электролиты.
Соли. Соли можно определить как электролиты, которые при растворении в воде диссоциируют, отщепляя положительные ионы, отличные от ионов водорода, и отрицательные ионы, отличные от гидроксид-ионов. Таких ионов, которые были бы общими для водных растворов всех солей, нет; поэтому соли и не обладают общими свойствами. Как правило, соли хорошо диссоциируют, и тем лучше, чем меньше заряды ионов, образующих соль.
При растворении кислых солей в растворе образуются катионы металла, сложные анионы кислотного остатка, а также ионы, являющиеся продуктами диссоциации этого сложного кислотного остатка, в том числе ионы Н+. Например, при растворении гидрокарбоната натрия диссоциация протекает согласно следующим уравнениям:
NaHCO3 = Na+ + HCO3-
HCO3- = Н+ + CO32-
При диссоциации основных солей образуются анионы кислоты и сложные катионы, состоящие из металла и гидроксогрупп. Эти сложные катионы также способны к диссоциации. Поэтому в растворе основной соли присутствуют ионы ОН-. Например, при растворении хлорида гидроксомагния диссоциации протекает согласно уравнениям:
MgOHCl = MgOH+ + Сl-
MgOH+ = Mg2+ + ОН-
Таким образом, теория электролитической диссоциации объясняет общие свойства кислот присутствием в их растворах ионов водорода, а общие свойства оснований — присутствием в их растворах гидроксид-ионов. Это объяснение не является, однако, общим. Известны химические реакции, протекающие с участием кислот и оснований, к которым теория электролитической диссоциации неприменима: В частности, кислоты и основания могут реагировать друг с другом, не будучи диссоциированы на ионы. Так, безводный хлороводород, состоящий только из молекул, легко реагирует с безводными основаниями. Кроме того, известны вещества, не имеющие в своем составе гидроксогрупп, но проявляющие свойства оснований. Например, аммиак взаимодействует с кислотами и образует соли (соли аммония), хотя в его составе нет групп ОН. Так, с хлороводородом он образует типичную соль — хлорид аммония:
NH3 + HC1 = NH4C1
Изучение подобного рода реакций, а также реакций, протекающих в неводных средах, привело к созданию более общих представлений о кислотах и основаниях. К важнейшим из современных теорий кислот и оснований принадлежит протонная теория, выдвинутая в 1923 г.
Согласно протонной теории, кислотой является донор протона, т. е. частица (молекула или ион), которая способна отдавать ион водорода протон, а основанием — акцептор протона, т. е. частица (молекула или ион), способная присоединять протон. Соотношение между кислотой и основанием определяется схемой:
Основание + Протон ↔ Кислота
Связанные этим соотношением основание и кислота называются сопряженными. Например, ион HSO4- является основанием, сопряженным кислоте H2SO4.
Реакцию между кислотой и основанием протонная теория представляет схемой:
(Кислота)1 + (Основание)2 = (Кислота)2 + (Основание)1
Например, в реакции
HC1 + NH3 = NH3+ + Сl-
ион Сl-— основание, сопряженное кислоте НС1, а ион NH3+ — кислота, сопряженная основанию NH3.
Существенным в протонной теории является то положение, что вещество проявляет себя как кислота или как основание в зависимости от того, с каким другим веществом оно вступает в реакцию. Важнейшим фактором при этом является энергия связи вещества с протоном. Так, в ряду NH3— Н2О— HF эта энергия максимальна для NH3 и минимальна для HF. Поэтому в смеси с NH3 вода функционирует как кислота, а в смеси с HF — как основание:
NH3 + Н2О = NH4+ + ОН-
HF + Н2О = F- + Н3О+
2.6 Ионно-молекулярные уравнения
При нейтрализации любой сильной кислоты любым сильным основанием на каждый моль образующейся воды выделяется около 57,6 кДж теплоты:
НСl + NaOH = NaCl + H2O + 57,53 кДж
НNO3 + КОН = КNO3 + H2O +57,61 кДж
Это говорит о том, что подобные реакции сводятся к одному процессу. Уравнение этого процесса мы получим, если рассмотрим подробнее одну из приведенных реакций, например, первую. Перепишем ее уравнение, записывая сильные электролиты в ионной форме, поскольку они существуют в растворе в виде ионов, а слабые— в молекулярной, поскольку они находятся в растворе преимущественно в виде молекул (вода — очень слабый электролит):
Н+ + Cl- + Na+ + ОН- = Na+ + Cl- + H2O
Рассматривая получившееся уравнение, видим, что в ходе реакции ионы Na+ и Cl- не претерпели изменений. Поэтому перепишем уравнение еще раз, исключив эти ионы из обеих частей уравнения. Получим:
Н+ + ОН- = H2O
Таким образом, реакции нейтрализации любой сильной кислоты любым сильным основанием сводятся к одному и тому же процессу - к образованию молекул воды из ионов водорода и гидроксид-ионов. Ясно, что тепловые эффекты этих реакций тоже должны быть одинаковы.
Строго говоря, реакция образования воды из ионов обратима, что можно выразить уравнением
Н+ + ОН- ↔ H2O
Однако, как мы увидим ниже, вода — очень слабый электролит, и диссоциирует лишь в ничтожно малой степени. Иначе говоря, равновесие между молекулами воды и ионами сильно смещено в сторону образования молекул. Поэтому практически реакция нейтрализации сильной кислоты сильным основанием протекает до конца.
При смешивании раствора какой-либо соли серебра с соляной кислотой или с раствором любой ее соли всегда образуется характерный белый творожистый осадок хлорида серебра:
AgNO3+ НС1 = AgCl↓ + HNO3
Ag2SO4 + CuCl2 = 2AgCl↓ + CuSO4
Подобные реакции также сводятся к одному процессу. Для того чтобы получить его ионно-молекулярное уравнение, перепишем, например, уравнение первой реакции, записывая сильные электролиты, как и в предыдущем примере, в ионной форме, а вещество, находящееся в осадке, в молекулярной:
Ag+ + NO3- + Н+ + С1- = AgCl↓+ Н+ + NO3-
Как видно, ионы Н+ и NO3- не претерпевают изменений в ходе реакции. Поэтому исключим их и перепишем уравнение еще раз:
Ag+ + С1- = AgCl↓
Это и есть ионно-молекулярное уравнение рассматриваемого процесса.
Здесь также надо иметь в виду, что осадок хлорида серебра находится в равновесии с ионами Ag +и С1- в растворе, так что процесс, выраженный последним уравнением, обратим:
Ag+ + С1- ↔ AgCl↓
Однако, вследствие малой растворимости хлорида серебра, это равновесие очень сильно смещено вправо. Поэтому можно считать, что реакция образования AgCl из ионов практически доходит до конца.
Образование осадка AgCl будет наблюдаться всегда, когда в одном растворе окажутся в значительной концентрации ионы Ag+и С1- . Поэтому с помощью ионов серебра можно обнаружить присутствие в растворе ионов С1- и, обратно, с помощью хлорид-ионов — присутствие ионов серебра; ион С1- может служить реактивом на ион Ag+ , а ион Ag+ — реактивом на ион С1.
В дальнейшем мы будем широко пользоваться ионно-молекулярной формой записи уравнений реакций с участием электролитов.
Для составления ионно-молекулярных уравнений надо знать, какие соли растворимы в воде и какие практически нерастворимы. Общая характеристика растворимости в воде важнейших солей приведена в табл.2.
Ионно-молекулярные уравнения помогают понять особенности протекания реакций между электролитами. Рассмотрим в качестве примера несколько реакций, протекающих с участием слабых кислот и оснований.
Таблица 2. Растворимость важнейших солей в воде
| Анионы и катионы | Растворимость солей |
|
NO3- Cl- SO42- CO32- PO43- Na+, K+, NН4+ |
Растворимы все соли Растворимы все соли, кроме AgCl, Cu Cl, PbCl2 и Hg2Cl2 Растворимы все соли, кроме BaSO4, SrSO4 и PbSO4; малорастворим СаSO4 Из средних солей растворимы только соли натрия, калия и аммония То же Растворимы почти все соли |
Как уже говорилось, нейтрализация любой сильной кислоты любым сильным основанием сопровождается одним и тем же тепловым эффектом, так как она сводится к одному и тому же процессу — образованию молекул воды из ионов водорода и гидроксид-иона. Однако при нейтрализации сильной кислоты слабым основанием, слабой кислоты сильным или слабым основанием тепловые эффекты различны. Напишем ионно-молекулярные уравнения подобных реакций.
Нейтрализация слабой кислоты (уксусной) сильным основанием (гидроксидом натрия):
СН3СООН + NaOH = CH3COONa + Н2О
Здесь сильные электролиты— гидроксид натрия и образующаяся соль, а слабые — кислота и вода:
СН3СООН + Na++ ОН- = СН3СОО- + Na+ + Н2О
Как видно, не претерпевают изменений в ходе реакции только ионы натрия. Поэтому ионно-молекулярное уравнение имеет вид:
СН3СООН + ОН- = СН3СОО- + Н2О
Нейтрализация сильной кислоты (азотной) слабым основанием (гидроксидом аммония):
HNO3 + NH4OH = NH4NO3 + Н2О
Здесь в виде ионов мы должны записать кислоту и образующуюся соль, а в виде молекул — гидроксид аммония и воду:
Н+ + NO3- + NH4OH = NH4- + NH3- + Н2О
Не претерпевают изменений ионы NO3- . Опуская их, получаем ионно-молекулярное уравнение:
Н+ + NH4OH= NH4+ + Н2О
Нейтрализация слабой кислоты (уксусной) слабым основанием (гидроксидом аммония):
СН3СООН + NH4OH = CH3COONH4 + Н2О
В этой реакции все вещества, кроме образующейся соли,— слабые электролиты. Поэтому ионно-молекулярная форма уравнения имеет вид:
СН3СООН + NH4OH =СН3СОО- + NH4+ + Н2О
Сравнивая между собой полученные ионно-молекулярные уравнения, видим, что все они различны. Поэтому понятно, что неодинаковы и теплоты рассмотренных реакций.
Реакции нейтрализации сильных кислот сильными основаниями, в ходе которых ионы водорода и гидроксид-ионы соединяются в молекулу воды, протекают практически до конца. Реакции же нейтрализации, в которых хотя бы одно из исходных веществ — слабый электролит и при которых молекулы малодиссоциирующих веществ имеются не только в правой, но и в левой части ионно-молекулярного уравнения, протекают не до конца. Они доходят до состояния равновесия, при котором соль сосуществует с кислотой и основанием, от которых она образована. Поэтому уравнения подобных реакций правильнее записывать как обратимые реакции:
СН3СООН + ОН- ↔ СН3СОО- + Н2О
Н+ + NH4OH↔ NH4+ + Н2О
СН3СООН + NH4OH ↔ СН3СОО- + NH4+ + Н2О
2.7 Произведение растворимости.
При растворении твердого тела в воде растворение прекращается, когда получается насыщенный раствор, т. е. когда между растворяемым веществом и находящимися в растворе молекулами того же вещества установится равновесие. При растворении электролита, например, соли, в раствор переходят не молекулы, а ионы; следовательно, и равновесие в насыщенном растворе устанавливается между твердой солью и перешедшими в раствор ионами. Например, в насыщенном растворе сульфата кальция устанавливается равновесие твердая ионы в соль растворе
CaSO4 ↔Са2+ + SO42-
Константа равновесия для этого процесса выразится уравнением:
К = [Са2+] [SO42-]
[CaSO4]
Знаменатель дроби — концентрация твердой соли — представляет собою постоянную величину, которую можно ввести в константу.
Тогда, обозначая
К = [CaSO4] = К’
получим
[Са2+] [SO42-] = К’
Таким образом, в насыщенном растворе электролита произведение концентраций его ионов есть величина постоянная при данной температуре. Эта величина количественно характеризует способность электролита растворяться; ее называют произведением растворимости электролита и обозначают буквами ПР.
Заменив величину К’ на ПРCaSO4, получим:
ПРCaSO4 = [Са2+] [SO42-]
Численное значение произведения растворимости электролита нетрудно найти, зная его растворимость. Например, растворимость сульфата кальция при 20°С равна 1,5∙10-2 моль/л. Это значит, что в насыщенном растворе концентрация каждого из ионов Са2+ и SO42- равна 1,5∙10-2 моль/л.
Следовательно, произведение растворимости этой соли
ПРCaSO4 = [Са2+] [SO42-] = (1,5∙10-2)2 = 2,25∙10-4
В тех случаях, когда электролит содержит два или несколько одинаковых ионов, концентрации этих ионов при вычислении произведения растворимости должны быть возведены в соответствующие степени. Например:
ПРРbCl2 = [Pb2+] [I-]2
Знание произведения растворимости позволяет решать вопросы, связанные с образованием или растворением осадков при химических реакциях, что особенно важно для аналитической химии. Надо, однако, иметь в виду, что произведение растворимости, вычисленное без учета коэффициентов активности, является постоянной величиной только для малорастворимых электролитов и при условии, что концентрации других находящихся в растворе ионов невелики. Это объясняется тем, что коэффициенты активности близки к единице только в очень разбавленных растворах. Для хорошо растворимых электролитов значение произведения концентраций ионов в насыщенном растворе может сильно изменяться в присутствии других веществ. Это происходит вследствие изменения коэффициентов активности ионов. Поэтому расчеты, производимые по произведению растворимости без учета коэффициентов активности, приводят в этих случаях к неверным результатам.
В табл.3 приведены величины произведения растворимости некоторых малорастворимых соединений в воде.
Таблица 3.
Произведение растворимости некоторых веществ при 25°С
| Соединение | Произведение растворимости | Соединение | Произведение растворимости |
|
AgCl AgBr AgI Cu(OH)2 Zn(OH)2 BaSO4 CaSO4 |
1,8∙10-10 6∙10-13 1∙10-16 2,2∙10-20 1∙10-17 1,1∙10-10 1,3∙10-4 |
CaCO3 CuS Cu2S FeS HgS MnS ZnS |
5∙10-9 6∙10-36 1∙10-48 5∙10-18 10-52 2,5∙10-10 10-23 |
2.8.Диссоциация воды. Водородный показатель
Чистая вода очень плохо проводит электрический ток, но все же обладает измеримой электропроводностью, которая объясняется небольшой диссоциацией воды на ионы водорода и гидроксид-ионы:
Н2О ↔ Н+ + ОН-
По величине электропроводности чистой воды можно вычислить концентрацию ионов водорода и гидроксид-ионов в воде. При 25°С она равна 10-7моль/л.
Напишем выражение для константы диссоциации воды:
К = [Н+] [ОН-]
[Н2О]
Перепишем это уравнение следующим образом:
[Н+] [ОН-] = [Н2О] К
Поскольку степень диссоциации воды очень мала, то концентрация недиссоциированных молекул Н2О в воде практически равна общей концентрации воды, т. е. 55,55 моль/л (1 л содержит 1000 г, воды, т. е.
100:18,02 = 55,55 молей). В разбавленных водных растворах концентрацию воды можно считать такой же. Поэтому, заменив в последнем уравнении произведение [Н2О] К новой константой КН2О, будем иметь:
[Н+] [ОН-] = КН2О
Полученное уравнение показывает, что для воды и разбавленных водных растворов при неизменной температуре произведение концентраций ионов водорода и гидроксид-ионов есть величина постоянная. Эта постоянная величина называется ионным произведением воды. Численное значение ее нетрудно получить, подставив в последнее уравнение концентрации ионов водорода и гидроксид-ионов. В чистой воде при 25°С [Н+] [ОН-] = 1∙10-7моль/л. Поэтому для указанной температуры:
КН2О = 10-7∙10-7 = 10-14
Растворы, в которых концентрации ионов водорода и гидроксид-ионов одинаковы, называются нейтральными растворами. При 25 °С, как уже сказано, в нейтральных растворах концентрация как ионов водорода, так и гидроксид-ионов равна10-7 моль/л. В кислых растворах больше концентрация ионов водорода, в щелочных— концентрация гидроксид-ионов. Но какова бы ни была реакция раствора, произведение концентраций ионов водорода и гидроксид-ионов остается постоянным.
Если например, к чистой воде добавить столько кислоты, чтобы концентрация ионов водорода повысилась до 10-3 моль/л, то концентрация гидроксид-ионов понизится так, что произведение [Н+] [ОН-] останется равным 10-14. Следовательно, в этом растворе концентрация гидроксид-ионов будет:
[ОН-] = 10-14/10-3 = 10-11моль/л
Наоборот, если добавить к воде щелочи и тем повысить концентрацию гидроксид-ионов, например, до 10-5моль/л, то концентрация ионов водорода составит:
[Н+] = 10-14/10-5 = 10-9моль/л
Эти примеры показывают, что если концентрация ионов водорода в водном растворе известна, то тем самым определена и концентрация гидроксид-ионов. Поэтому как степень кислотности, так и степень щелочности раствора можно количественно охарактеризовать концентрацией ионов водорода:
Нейтральный раствор [Н+] = 10-7моль/л
Кислый [Н+] > 10-7моль/л
Щелочной [Н+] <10-7моль/л
Кислотность или щелочность раствора можно выразить другим, более удобным способом: вместо концентрации ионов водорода указывают ее десятичный логарифм, взятый с обратным знаком. Последняя величина называется водородным показателем и обозначается через рН:
рН = -lg[Н+]
Например, если [Н+]= 10-5моль/л, то рН = 5; если [Н+]= 10-9моль/л, то рН = 9 и т. д. Отсюда ясно, что в нейтральном растворе ([Н+]= 10-7моль/л) рН = 7. В кислых растворах рН < 7, и тем меньше, чем кислее раствор. Наоборот, в щелочных растворах рН >7, и тем больше, чем больше щелочность раствора.
Для измерения рН существуют различные методы. Приближенно реакцию раствора можно определить с помощью специальных реактивов, называемых индикаторами, окраска которых меняется в зависимости от концентрации ионов водорода. Наиболее распространенные индикаторы — метиловый оранжевый, метиловый красный, фенолфталеин. В табл.4 дана характеристика некоторых индикаторов.
Таблица 4. Важнейшие индикаторы
| Название индикатора | Цвет индикатора в различных средах | ||
| в кислой | в нейтральной | в щелочной | |
|
Метиловый оранжевый Метиловый красный Фенолфталеин Лакмус |
Красный (рН < 3,1) Красный (рН < 4,2) Бесцветный (рН < 8,0) Красный (рН < 5) |
Оранжевый (3,1 < рН < 4,4) Оранжевый (4,2 < рН < 6,3) Бледно-малинов. (8,0 < рН < 9,8) Фиолетовый (5 < рН < 8) |
Желтый (рН > 4,4) Желтый (рН > 6,3) Малинов. (рН > 9,8) Синий (рН > 8) |
Для многих процессов величина рН имеет большое значение. Так, рН крови человека и животных имеет строго постоянное значение. Растения могут нормально произрастать лишь при значениях рН почвенного раствора, лежащих в определенном интервале, характерном для данного вида растения. Свойства природных вод, в частности их коррозионная активность, сильно зависят от их рН.
2.9 Смещение ионных равновесий
Равновесие в растворах электролитов, как и всякое химическое равновесие, сохраняется неизменным, пока определяющие его условия не меняются; изменение условий влечет за собой нарушение равновесия.
Так, равновесие нарушается при изменении концентрации одного из участвующих в этом равновесии ионов: при ее увеличении происходит процесс, в ходе которого эти ионы связываются. Например, если в раствор уксусной кислоты, диссоциирующей согласно уравнению ввести какую-либо соль этой кислоты и тем самым увеличить концентрацию ионов СН3СОО-, то, в соответствии с принципом Ле Шателье, равновесие смещается влево, т. е. степень диссоциации уксусной кислоты уменьшается.
СН3СООН ↔ СН3СОО- + H+
Отсюда следует, что введение в раствор слабого электролита одноименных ионов (т. е. ионов, одинаковых с одним из ионов электролита) уменьшает степень диссоциации этого электролита. Наоборот, уменьшение концентрации одного из ионов вызывает диссоциацию нового количества молекул. Например, при введении в раствор указанной кислоты гидроксид-ионов, связывающих ионы водорода, диссоциация кислоты возрастает.
Аналогично нарушается равновесие в случае малорастворимого электролита: всякий раз, как только произведение концентраций ионов малорастворимого электролита в растворе превысит, величину произведения растворимости, образуется осадок. Так, если к насыщенному раствору сульфата кальция добавить другой, хорошо растворимый электролит, содержащий общий с сульфатом кальция ион, например, сульфат калия, то вследствие увеличения концентрации ионов SO42- равновесие сместится в сторону образования кристаллов CaSО4; ионы Са2+ и SO42- будут удаляться из раствора, образуя осадок. Процесс будет идти до тех пор, пока произведение концентраций этих ионов станет равно произведению растворимости CaSO4. В итоге количество сульфата кальция в растворе уменьшится.
Таким образом, растворимость электролита уменьшается от введения в раствор одноименных ионов. Исключением являются те случаи, когда происходит связывание одного из находящихся в растворе ионов с вводимыми ионами в более сложные (комплексные) ионы.
На основании рассмотренных примеров можно сделать общий вывод.
Обязательным условием течения реакций между электролитами является удаление из раствора тех или иных ионов — например, вследствие образования слабо диссоциирующих веществ, или веществ, выделяющихся из раствора в виде осадка или газа. Иначе говоря, реакции в растворах электролитов всегда идут в сторону образования наименее диссоциированных или наименее растворимых веществ.
Из этого, в частности, следует, что сильные кислоты вытесняют слабые из растворов их солей. Например, при взаимодействии ацетата натрия с соляной кислотой реакция практически нацело протекает с образованием уксусной кислоты.
CH3COONa + НС1 = СН3СООН + NaCl
или в ионно-молекулярной форме:
СН3СОО- + H+ = СН3СООН
Аналогично протекают реакции между сильными основаниями и солями слабых оснований. Например, при действии гидроксида натрия на сульфат железа (П) выделяется гидроксид железа(II)
FeSO4+ 2NaOH = Na2SO4+ Fe(OH)2↓
или в ионно-молекулярной форме:
Fe2+ + 2OH- = Fe(OH)2↓
Последняя реакция служит примером образования не только слабого, но и малорастворимого электролита.
С рассмотренной точки зрения становится ясным различие между реакциями нейтрализации сильной кислоты сильным основанием и случаями нейтрализации, когда хотя бы одно из исходных веществ — слабый электролит. При нейтрализации сильной кислоты сильным основанием в растворе образуется только один слабый электролит — вода. При этом равновесие сильно смещено вправо и реакция в этом случае доходит практически до конца
Н+ + OH- ↔ Н2О
При нейтрализации же слабой кислоты или слабого основания в растворе существуют, по крайней мере, два слабых электролита — вода и слабая кислота или слабое основание. Например, при нейтрализации уксусной кислоты сильным основанием в растворе устанавливаются два равновесия:
Н+ + СН3СОО- ↔ СН3СООН
Н+ + ОН- ↔ Н2О
Ион водорода может, таким образом, связаться в молекулу уксусной кислоты или в молекулу воды. Ионы СН3СОО- и ОН- как бы «конкурируют» друг с другом в связывании иона водорода. Поэтому в данном случае реакция нейтрализации доходит не до конца, а до состояния равновесия:
СН3СООН + OH- ↔ СН3СОО- + Н2О
Однако это равновесие сильно смещено вправо, поскольку вода значительно более слабый электролит, чем уксусная кислота, так что связывание ионов Н+ в молекулы воды происходит полнее, чем в молекулы уксусной кислоты.
При нейтрализации слабого основания — гидроксида аммония - сильной кислотой в растворе тоже устанавливаются два равновесия:
NH4+ + OH- ↔ NH4OH
H+ + OH- ↔ Н2О
Здесь конкурируют ионы NH4+ и H+, связывающие гидроксид-ионы в недиссоциированные молекулы. В результате и эта реакция доходит не до конца, а до состояния равновесия:
H+ + NH4OH ↔ NH4+ + Н2О
Но поскольку вода — значительно более слабый электролит, чем NH4OH, равновесие сильно смещено вправо.
Подобные процессы происходят и при реакциях, в ходе которых малорастворимое вещество превращается в растворимый, но слабо диссоциирующий продукт. К таким реакциям относится, например, растворение сульфидов некоторых металлов в соляной кислоте. Так, взаимодействие сульфида марганца с соляной кислотой выражается уравнением
MnS(к) + 2НСl =MnCl2 + H2S
или
MnS(к) + 2Н+ =Mn2+ + H2S
Присутствие в числе исходных веществ малорастворимого электролита (MnS), при образовании которого связываются ионы S2-, обусловливает протекание реакции влево. С другой стороны, при образовании слабого электролита (H2S) также связываются ионы S2-, что способствует протеканию реакции вправо. Таким образом, ионы S2- участвуют в двух конкурирующих процессах, приводящих к установлению двух равновесий:
S2- + Mn2+ ↔ MnS(к)
и
S2- + 2Н+ ↔ H2S
Направление рассматриваемой реакции зависит от того, какое из двух веществ — H2S или MnS — в большей степени связывает ионы S2-. Суммарная константа диссоциации сероводорода К = К1 К2 = 6∙10-22; произведение же растворимости MnS равно 2,5∙10-10 . Отсюда ясно, что связывание ионов S2- в молекулы сероводорода происходит полнее, чем в MnS. Поэтому рассматриваемая реакция протекает вправо — сульфид марганца растворяется в соляной кислоте.
Аналогичные два равновесия устанавливаются в системе соляная кислота — сульфид меди(II). Но произведение растворимости CuS очень мало, оно равно 6∙10-36. Поэтому связывание ионов S2- в CuS происходит полнее, чем в молекулы сероводорода, и равновесие в системе
CuS(к) + 2HC1 ↔ CuCl2 + Н2S
смещено влево; сульфид меди(II) нерастворим в соляной кислоте.
Рассмотренные закономерности позволяют понять поведение амфотерных гидроксидов. Так, равновесие между осадком гидроксида цинка и раствором нарушается при добавлении как кислоты, так и щелочи. В этом случае равновесие можно записать в форме:
2Н+ + ZnO22- ↔ Zn(OH)2 ↔ Zn2+ + 2OH-
раствор
↕
Zn(OH)2
осадок
При добавлении к гидроксиду цинка кислоты возрастает концентрация ионов водорода. Произведение [Н+][ОН-] становится больше ионного произведения воды — идет процесс образования молекул Н2О из ионов; при этом нарушается равновесие и в системе Zn(OH)2. Согласно принципу Ле Шателье, вследствие возрастания концентрации ионов Н+ и расхода ионов ОН-, диссоциация Zn(OH)2 по типу кислоты подавляется, а по типу основания усиливается. В итоге осадок Zn(OH)2 растворяется и образуется соль, в которой цинк является катионом. Например, в случае соляной кислоты пойдет реакция:
Zn(OH)2+ 2HC1= ZnСl2 + H2O
При добавлении к гидроксиду цинка щелочи возрастает концентрация ионов ОН-: в этом случае процесс идет в направлении связывания ионов водорода. Равновесие в системе нарушается, но теперь преобладает диссоциация Zn(OH)2 по типу кислоты. В итоге осадок Zn(OH)2 растворяется и образуется соль, в которой цинк входит в состав аниона. Например, при добавлении NaOH идет реакция:
Zn(OH)2 + 2NaOH = Na2ZnО2 + 2H2O
В обоих случаях процесс возможен и протекает потому, что связывание ионов Н+ и ОН- в молекулы воды происходит в большей степени, чем в молекулы Zn(OH)2.
2.10 Гидролиз солей
Гидролизом называется взаимодействие вещества с водой, при котором составные части вещества соединяются с составными частями воды. Примером гидролиза может служить взаимодействие хлорида фосфора(III) РС13 с водой. В результате этой реакции образуются фосфористая кислота Н3РО3 и соляная кислота:
РС13 + ЗН2О = Н3РО3 + ЗНС1
Гидролизу подвержены соединения различных классов. Мы рассмотрим один из важнейших его случаев — гидролиз солей.
В случае реакций нейтрализации, в которых участвуют слабые кислоты и основания, реакции протекают не до конца. Значит при этом в той или иной степени протекает и обратная реакция (взаимодействие соли с водой), приводящая к образованию кислоты и основания. Это и есть гидролиз соли.
В реакции гидролиза вступают соли, образованные слабой кислотой и слабым основанием, или слабой кислотой и сильным основанием, или слабым основанием и сильной кислотой. Соли, образованные сильной кислотой и сильным основанием, гидролизу не подвергаются; нейтрализация в этом случае сводится к процессу
Н++ ОН- = Н2О
А обратная реакция — диссоциация молекулы воды на ионы протекает в ничтожно малой степени.
Рассмотрим гидролиз соли, образованной одноосновной кислотой и одновалентным металлом. В качестве примера возьмем ацетат натрия — соль слабой кислоты и сильного основания. Уравнение гидролиза этой соли имеет вид
CH3COONa + Н2О ↔ СН3СООН + NaOH
или
CH3COO- + Н2О ↔ СН3СООН + ОН-
Уравнение показывает, что в данном случае гидролизу подвергается анион соли и что реакция сопровождается образованием ионов ОН-. Но поскольку ионное произведение воды [Н+] [ОН-] величина постоянная, то при накоплении ионов ОН- концентрация ионов водорода уменьшается. Следовательно, растворы солей, образованных слабой кислотой и сильным основанием, имеют щелочную реакцию.
Аналогично в случае соли, образованной слабым основанием и сильной кислотой, гидролизу подвергается катион соли и реакция сопровождается образованием ионов Н+, например
NH4C1 + Н2О ↔ NH4OH + НС1
или
NH4+ + Н2О ↔ NH4OH + Н+
Накопление ионов Н+ приводит к уменьшению концентрации ионов ОН+. Таким образом, растворы солей, образованных слабым основанием и сильной кислотой, имеют кислую реакцию.
В рассмотренных случаях гидролизу подвергается не все количество находящейся в растворе соли, а только часть его. Иначе говоря, в растворе устанавливается равновесие между солью и образующими ее кислотой и основанием. Доля вещества, подвергающаяся гидролизу, — степень гидролиза зависит от константы этого равновесия, а также от температуры и от концентрации соли.
Запишем уравнение гидролиза в общем виде. Пусть НА — кислота, МОН — основание, МА — образованная ими соль. Тогда уравнение гидролиза будет иметь вид:
МА + Н2О ↔ НА + МОН
Этому равновесию отвечает константа:
К = [НА] [МОН]
[МА] [Н2О]
Концентрация воды в разбавленных растворах представляет собою практически постоянную величину. Обозначая
К[Н2О] = Кг
получим:
К = [НА] [МОН]
[МА]
Величина Кг называется константой гидролиза соли. Ее значение характеризует способность данной соли подвергаться гидролизу; чем больше Кг, тем в большей степени (при одинаковых температуре и концентрации соли) протекает гидролиз.
Для случая соли, образованной слабой кислотой и сильным основанием, константа гидролиза связана с константой диссоциации кислоты Ккисл зависимостью:
Кг = КН2О
Ккисл
Это уравнение показывает, что Кг тем больше, чем меньше Ккисл. Иными словами, чем слабее кислота, тем в большей степени подвергаются гидролизу ее соли.
Для солей, образованных слабым основанием и сильной кислотой, аналогичное соотношение связывает константу гидролиза константой диссоциации основания Kосн:
Кг = КН2О
Косн
Поэтому, чем слабее основание, тем в большей степени подвергаются гидролизу образованные им соли.
Для солей, образованных слабой кислотой и слабым основанием, константа гидролиза связана с константами диссоциации кислоты и основания следующим соотношением:
Кг = КН2О
КкислКосн
Степень гидролиза определяется природой соли, ее концентрацией и температурой. Природа соли проявляется в величине константы гидролиза. Зависимость от концентрации выражается в том, что с разбавлением раствора степень гидролиза увеличивается. В самом деле, пусть мы имеем, например, раствор цианида калия. В нем устанавливается равновесие
КСN + Н2О ↔ HCN + KOH
которому отвечает константа
Кг = [НСN] [KOH]
[KCN]
Разбавим раствор в 10 раз. В первый момент концентрации всех веществ —KCN, HCN и КОН—уменьшаются в 10 раз. Вследствие этого числитель правой части уравнения константы гидролиза уменьшится в 100 раз, а знаменатель только в 10 раз. Но константа гидролиза, как всякая константа равновесия, не зависит от концентраций веществ. Поэтому равновесие в растворе нарушится. Для того чтобы оно вновь установилось, числитель дроби должен возрасти, а знаменатель — уменьшиться, т.е. некоторое количество соли должно дополнительно гидролизоваться. В результате этого концентрации HCN и КОН возрастут, а концентрация
KCN — уменьшится. Таким образом, степень гидролиза соли увеличится.
Влияние температуры на степень гидролиза вытекает из принципа Ле Шателье. Все реакции нейтрализации протекают с выделением теплоты, а гидролиз с поглощением теплоты. Поскольку выход эндотермических реакций с ростом температуры увеличивается, то и степень гидролиза растет с повышением температуры.
Из сказанного ясно, что для ослабления гидролиза растворы следует хранить концентрированными и при низких температурах. Кроме того, подавлению гидролиза способствует подкисление (в случае солей, образованных сильной кислотой и, слабым основанием) или подщелачивание (для солей, образованных сильным основанием и слабой кислотой) раствора.
Рассмотрим теперь гидролиз солей, образованных слабой многоосновной кислотой или слабым основанием многовалентного металла. Гидролиз таких солей протекает ступенчато. Так, первая ступень гидролиза карбоната натрия протекает согласно уравнению
Na2CO3 + H2O ↔ NaHCO3 + NaOH
или в ионно-молекулярной форме:
CO32- + H2O ↔ HCO3- + OH+
Образовавшаяся кислая соль в свою очередь подвергается гидролизу (вторая ступень гидролиза)
NaНCO3+ H2O ↔ H2CO3 + NaOH
или
НCO3- + H2O ↔ H2CO3 + OH-
Как видно, при гидролизе по первой ступени образуется ион
НCO3-,диссоциация которого характеризуется второй константой диссоциации угольной кислоты, а при гидролизе по второй ступени образуется угольная кислота, диссоциацию которой характеризует первая константа ее диссоциации. Поэтому константа гидролиза по первой ступени Кг,1, связана со второй константой диссоциации кислоты, а константа гидролиза по второй ступени Кг,2— с первой константой диссоциации кислоты. Эта связь выражается соотношениями:
Кг = КН2О
К2, кисл
Кг = КН2О
К-1, кисл
Поскольку первая константа диссоциации кислоты всегда больше второй, то константа гидролиза по первой ступени всегда больше, чем константа гидролиза по второй ступени:
Кг,1> Кг,1
По этой причине гидролиз по первой ступени всегда протекает в большей степени, чем по второй. Кроме того, ионы, образующиеся при гидролизе по первой ступени (в рассмотренном примере— ионы ОН-), способствуют смещению равновесия второй ступени влево, т. е. также подавляют гидролиз по второй ступени.
Аналогично проходит гидролиз солей, образованных слабый основанием многовалентного металла. Например, гидролиз хлорида меди (II) протекает по первой ступени с образованием хлорида гидроксомеди
СuCl2 + H2O ↔ CuOHCl + HCl
или в ионно-молекулярной форме:
Cu2+ + H2O ↔ CuOH+ + H+
Гидролиз по второй ступени происходит в ничтожно малой степени:
CuОН + + H2O ↔ Cu(OH)2 + H+
Протонная теория кислот и оснований рассматривает гидролиз как частный случай кислотно-основного равновесия: протон переходит от молекулы воды к данному иону или от данного иона к молекуле воды. Например, гидролиз аммония можно выразить уравнением:
NH4+ + H2O ↔ H3O+
+ NH3 ![]()
3. Применение электролитов
Электролиты играют важную роль в науке и технике. Они участвуют в электрохимических и многих биологических процессах, являются средой для органического и неорганического синтеза и электрохимического производства.
Устройства с твердыми оксидными электролитами. Главное предназначение твердых оксидных электролитов виделось в создании топливных элементов - химических источников тока, в которых энергия газа непосредственно превращается в электрическую. Топливные элементы - близкие родственники гальванических элементов. Но те служат, пока в их электролите и электродах есть активные вещества, а топливные элементы могут работать сколь угодно долго, пока к ним подводится горючее. Систематические исследования твердых оксидных электролитов начались в Германии в начале 50-х годов, а с конца 50-х развернулись в СССР, США и Канаде. В нашей стране эти работы с самого начала вел Институт химии Уральского филиала АН СССР (Свердловск, ныне Екатеринбург), и школа высокотемпературной электрохимии твердых электролитов, созданная на Урале, стала уникальной по широте охвата проблемы и глубине ее изучения.
Конструкций, в основе которых лежат твердые оксидные электролиты, запатентовано очень много, но принцип их действия одинаков и довольно прост. Это пробирка с парой электродов на стенке, снаружи и внутри. Она помещена в нагреватель; внутрь пробирки и в пространство, ее окружающее, можно подводить газ. Посмотрим, какие функции могут выполнять такие устройства.
Потенциометрические датчики состава газа. Наверное, они наиболее просты. Электроды в разных газах приобретают разные потенциалы. Если, скажем, внутри пробирки находится чистый кислород, а снаружи - газ с неизвестной его концентрацией, то по разности потенциалов электродов можно эту концентрацию определить.
Потенциометрические датчики позволяют определять состав и более сложных газовых смесей, содержащих углекислый и угарный газы, водород и водяной пар. Если стерженек из твердого электролита с электродами на торцах нагрет неравномерно, он начнет терять кислород и между электродами возникнет разность потенциалов. По ее величине можно определить, например, состав выхлопных газов автомобильного двигателя. На Западе, где требования к чистоте выхлопных газов очень строги, такие датчики выпускаются миллионами. У нас же на такие "пустяки" пока не обращают внимания.
Кислородные датчики пока
единственные устройства с твердыми оксидными электролитами, нашедшие
практическое применение.![]()
Кислородные насосы. Пусть во внешнее пространство пробирки подается воздух или газ, содержащий кислород. Если внешний электрод стал анодом, а внутренний - катодом, то из газа в пробирку пойдет чистый кислород. Подобные устройства - кислородные насосы - могут найти применение там, где потребление кислорода невелико или требуется его высокая чистота.
В медицине, например,
используется и чистый кислород, и воздух с пониженным содержанием кислорода -
так называемая "гипоксическая смесь", или "горный воздух".
Электрохимические насосы наряду с мембранными оксигенаторами позволят решить
массу проблем, особенно в медицинских учреждениях, удаленных от промышленных
центров. В атмосфере с пониженным содержанием кислорода значительно дольше
хранятся продукты питания, и устройства с кислородными насосами могут стать
экономичней привычных холодильников.![]()
Электролизеры. Теперь к внешнему электроду - катоду - подводят водяной пар или углекислый газ. На катоде будет происходить разложение пара или углекислого газа, а на аноде в обоих случаях выделяется кислород. Уникальная способность этого высокотемпературного электролизера одновременно разлагать водяной пар и углекислый газ позволяет создать систему жизнеобеспечения, скажем, на космических объектах.
Теплоэлектрогенераторы. Человек сделал первый шаг к независимости от природы, научившись сохранять огонь, поистине универсальный источник энергии. Костер давал тепло и свет, на нем готовили пищу, он расходовал ровно столько топлива, сколько было необходимо. Костер тысячелетиями оставался главной энергетической установкой человека, и неудивительно, что мы испытываем какую-то ностальгию по очагу с горящими дровами.
Еще в конце прошлого века свет давали свечи и керосиновые лампы, а тепло - печи. Лишь немногим более ста лет назад на человека начало работать электричество, которое могло давать свет, тепло, механическую работу. Одно время казалось, что достаточно подвести к жилищу только электрическую энергию, а уж там преобразовывать ее во что угодно. Но сказала свое слово экономика: кпд электростанции менее 40%, потери при передаче и обратном превращении электричества в другие виды энергии тоже значительны. Ясно, что там, где нужно только тепло, его целесообразно получать прямо из топлива. И не случайно сегодня обсуждается простая идея: вернуть "очаг" в дом в виде электрохимического генератора с топливным элементом, преобразующим энергию топлива в электричество и тепло.
Топливные элементы. Пусть к внешним стенкам пробирки подается водород, а внутрь ее - кислород. Между электродами возникнет напряжение около вольта, по соединяющей их цепи потечет ток, и на электродах пойдут реакции, обратные тем, что проходят в электролизере. Внешний электрод станет анодом, внутренний - катодом, а устройство превратится в источник тока - твердооксидный топливный элемент.
Одно и то же устройство может служить и топливным элементом, и электролизером, позволяя аккумулировать электрическую энергию. В период низкого ее потребления невостребованная мощность электростанций используется для получения водорода. В пике потребления электролизер начинает работать как топливный элемент, производя электричество из водорода превратить уголь, нефть, различные газы и спирты (которые, например, в Бразилии используют как горючее для автомобилей). Элемент послужит основой электрохимического генератора, способного существенно изменить концепцию снабжения жилища энергией. Наиболее прост в техническом отношении генератор на природном газе - метане или пропане.
Как показывают исследования, его электрический кпд достигает 70%. Остальные 30% энергии топлива выделяются в виде тепла, которое можно использовать в паровых турбинах. Кпд такой комбинированной установки способно превысить 80% - столь высокой эффективности нет ни у одного генератора.
Восемь лет назад в Институте высокотемпературной электрохимии Уральского отделения РАН был изготовлен демонстрационный генератор на метане мощностью один киловатт. Но до практической реализации дело никак не дойдет. Опытно-конструкторские работы, которые уже начинались, до конца так и не доведены. Задача очень сложна, ее необходимо решать в рамках национальной программы, попытки разработать которую оказались пока безуспешными.
Электролит щелочной натриево-литиевый широко применяется в автомобильной и горно-добывающей промышленностях. Главное назначение этого электролита – заполнение различных щелочных аккумуляторов. Его используют для наполнения аккумуляторов электрических погрузчиков и специальных шахтных электровозов.
Электролит кислотный применяется для заливки в свинцовые аккумуляторы легкового и грузового автотранспорта.
Для приготовления электролита в ванну, футерованную свинцом, наливают плавиковую кислоту HF и в нее добавляют борную кислоту Н3ВО3. Полученную борфтористоводородную кислоту HBF4 фильтруют и, растворяют в ней углекислый кадмий. Для получения блестящего покрытия применяют электролит следующего состава, г/л. Покрытие производят при катодной плотности тока 9—10 А/дм2 и температуре 50°С.
При кадмировании деталей сложной геометрической формы применяют аммиакатные электролиты, рассеивающая способность которых выше, чем кислых. Чаще всего применяется электролит следующего состава, г/л.
Покрытие проводят при катодной плотности тока 0,5 — 1,0 А/дм2, рН=6,9 и температуре ванны 20—25°С. Этот электролит обладает хорошей буферной емкостью и не требует частых корректировок.
С введением декстрина улучшается структура поверхности и повышается катодная поляризация. Введение флюоресцина способствует получению мелкокристаллической структуры.
Цианистые электролиты позволяют получать покрытия очень высокого качества, однако в силу высокой токсичности компонентов и необходимости применять дорогие и сложные очистные сооружения для очистки сбрасываемых сточных вод эти электролиты на светотехнических заводах не применяют.
Другие электролиты, такие, как фенолсульфатные и этилендиаминовые, не получили широкого применения, так как работа с ними малопроизводительна.
Пассивирование кадмиевых покрытий производят значительно реже, чем цинковые.
При пассивировании детали
погружают на 5—10 с в раствор, после чего их вынимают и тщательно промывают в
проточной воде, сушат детали в потоке теплого воздуха.![]()
Заключение
Итак, подведем итоги: Электролитами называют вещества, растворы и сплавы которых с другими веществами электролитически проводят гальванический ток.
Признаком электролитической проводимости в отличии от металлической должно считать возможность наблюдать химическое разложение данного вещества при более или менее продолжительном прохождении тока. В химически чистом состоянии электролиты обыкновенно обладают ничтожно малой электропроводностью.
Термин электролит введен в науку Фарадеем. К.Э. К электролитам до самого последнего времени относили типичные соли, кислоты и щелочи, а также воду. Исследования неводных растворов, а также исследования при очень высоких температурах значительно расширили эту область.
И.А. Каблуков, Кади, Карара, П. И. Вальден и др. показали, что не только водные и спиртовые растворы заметно проводят ток, но также растворы в целом ряде других веществ, как например, в жидком аммиаке, жидком сернистом ангидриде и т. п.
Найдено также, что многие вещества и смеси их, превосходные изоляторы при обыкновенной температуре как например, безводные окислы металлов (окись кальция, магния и др.), при повышении температуры становятся электролитическими проводниками.
Известная лампа накаливания Нернста, принцип которой был открыт гениальным Яблочковым, представляет превосходную иллюстрацию этих фактов. Смесь окислов — “тельце для накаливания” в лампе Нернста, не проводящая при обыкновенной температуре при 700° делается превосходным и притом сохраняющим твердое состояние электролитическим проводником.
И напоследок считаю нужным сказать об определении электролитов, данном маститым Гитторфом пятьдесят лет тому назад: “Электролиты — это соли”. Этим определением Гитторф частью предвосхитил современную теорию электролитической диссоциации, указав на то, что типичное свойство солей, которое мы теперь определяем, как способность к электролитической диссоциации, должно быть признаком всякого электролита.
Список использованной литературы
1. Глинка Н.Л. Задачи и упражнения по общей химии. Учебное пособие для вузов./ Под ред. В.А. Рабиновича и Х. М. Рубиной. – 21-е изд., стереотипное – Л.: Химия, 1981.
2. Глинка Н.Л. Общая химия: Учебное пособие для вузов. – 22–е издание., испр./Под ред. Рабиновича В.А. – Л.: Химия, 1982.
3. Глинка Н.Л. Общая химия: Учебник для нехимических вузов. – М.: Химия, 1988.
4. Журнал "Наука и жизнь" № 2, 1999 г.
5. Лавриенко В.Н., В.П. Ратников, В.Ф. Голубь и др. Концепции современного естествознания: Учебник для вузов./ Под ред. проф. В.Н. Лавриенко, проф. В.П. Ратникова. – М.: Культура и спорт, ЮНИТИ,1977.
6. Ландсберг Г.С. Элементарный учебник физики: Учебное пособие. В 3-х т./ Под ред. Г.С. Ландсберга. Т. II. Электричество и магнетизм. – 10-е изд., перераб. – М.: Наука. Главная редакция физико-математической литературы, 1985.
7. Электрохимия расплавленных солевых и твердых электролитов. Термодинамика солевых и окисных систем. Свердловск., 1969г.
© 2009 База Рефератов