
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Курсовая работа: Рвотные и противорвотные препараты
Курсовая работа: Рвотные и противорвотные препараты
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ УКРАИНЫ
ГОСУДАРСТВЕННОЕ ВЫСШЕЕ УЧЕБНОЕ ЗАВЕДЕНИЕ
УКРАИНСКИЙ ГОСУДАРСТВЕННЫЙ
ХИМИКО-ТЕХНОЛОГИЧЕСКИЙ УНИВЕРСИТЕТ
Кафедра технологии органических веществ и фармацевтических препаратов
КУРСОВАЯ РАБОТА
по курсу: «Технология биологически активных веществ»
на тему:
«РВОТНЫЕ И ПРОТИВОРВОТНЫЕ ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ»
выполнила
ст. гр. 4-Ф-62
________________Лесунова И.А.
(подпись)
проверил
доц. каф. ТОРФП
________________Ничволода В.М.
(подпись)
Днепропетровск
2008 г.
ОГЛАВЛЕНИЕ
Введение. 3
1. Нормальная и патологическая физиология. 4
1.1 Нормальная физиология. 4
1.2 Патологическая физиология. 5
2. История открытия рвотных и противорвотных лекарственных препаратов 9
3. Классификация рвотных и противорвотных лекарственных препаратов. 12
3.1 Классификация препаратов по Машковскому. 12
3.2 Классификация по действию на нейромедиаторные процессы.. 13
4. Механизм биологической активности рвотных и противорвотных лекарственных средств. 14
4.1 Лекарственные средства, блокирующие серотониновые рецепторы.. 14
4.2 Лекарственные средства, блокирующие дофаминовые рецепторы.. 15
4.3 Препараты, блокаторующие гистаминовые Н1-рецепторы.. 17
4.4 М-холиноблокаторы.. 18
5. Методы получения рвотных и противорвотных лекарственных средств. 19
5.1 Синтез рвотных лекарственных средств. 19
5.1.1 Синтез апоморфина. 19
5.1.2 Синтез О, О´-диацилпроизводных апоморфина. 19
5.1.3 Синтез бромокриптина. 21
5.2 Синтез противорвотных лекарственных средств. 23
5.2.1 Синтез аминазина. 23
5.2.2 Синтез анестезина. 26
5.2.4 Синтез бромкамфоры.. 29
5.2.4 Синтез валидола. 30
6. Методы анализа рвотных и противорвотных лекарственных средств. 32
6.1 Методы анализа рвотных лекарственных средств. Анализ апоморфина. 32
6.2 Методы анализа противорвотных лекарственных средств. 32
6.2.1 Анализ аминазина. 32
6.2.2 Анализ анестезина. 34
6.2.3 Анализ бромкамфоры.. 34
6.2.4 Анализ валидола. 35
7. Сводная таблица препаратов. 37
7.1 Рвотные лекарственные препараты.. 37
7.2 Противорвотные лекарственные препараты.. 38
Словарь терминов. 49
Литература. 54
Рвотные средства лекарственные вещества, вызывающие рвоту [1]. К ним относится наиболее широко используемый апоморфин. Рефлекторно вызывают рвоту сульфаты меди и цинка, в больших дозах препараты термопсиса (Thermopsis lanceolata семейства бобовых) и ипекакуаны (Cephaelis ipecacuanha Uragoga ipecacuanha семейства мареновых). Сердечные гликозиды могут вызывать рвоту (при передозировке). Рвотные средства применяют для ускорения рвоты при попадании в желудок раздражающих и токсичных веществ.
Противорвотные средства успокаивают рвоту [2]. Такими свойствами обладают холинолитические, антигистаминные и особенно нейролептические средства.
Весьма эффективными противорвотными средствами являются нейролептические препараты группы фенотиазина и бутирофенона, действующие на дофаминергические системы. Высокой противорвотной активностью в ряду производных фенотиазина обладают этаперазин, метеразин, трифтазин, фторфеназин и др., в ряду бутирофенонов – галоперидол и др.
Если рвота вызвана местным раздражением желудка, то после удаления раздражающих веществ в случае необходимости могут быть применены обволакивающие и вяжущие средства.
Уменьшение возбудимости рецепторов желудка и подавление тошноты и рвоты может быть достигнуто назначением местных анестетиков (анестезин, новокаин).
Производные марихуаны, включая собственно тетрагидроканнабинол (дронабинол), являются эффективными противорвотными средствами для некоторых пациентов, в том числе для тех, кому не помогают другие препараты.
Для снятия возбуждения рвотного центра ранее основное применение имели седативные и снотворные средства [1].
Для этих же целей широко применяются противогистаминные препараты: димедрол, дипразин и др. [2]. Антигистаминные, дименгидринат и прометазина гидрохлорид эффективны при тошноте, вызванной поражением внутреннего уха.
В связи с комплексностью нейрохимической организации рвотного акта наиболее выраженный противорвотный эффект может наблюдаться при комбинированном применении веществ, влияющих на разные медиаторные системы (например, нейролептиков и противогистаминных препаратов и др.).
Антагонистами дофаминовых рецепторов являются метоклопрамид, домперидон, диметпрамид, также применяемые в качестве противорвотных средств [1]. Селективные антагонисты допамина (например, метоклопрамид) могут с большим успехом чем фенотиазины применяться для устранения выраженных тошноты и рвоты [2].
Самыми новыми препаратами для лечения тошноты и рвоты являются антагонисты серотонина [1]. Ондансетрон относится к антагонистам 5-НТ3-рецепторов, которые являются одними из трех видов серотониновых рецепторов, выделенных в настоящее время. Эти рецепторы имеются как в ЦНС, так и в желудочно-кишечном тракте.
Ондансетрон существует только в форме для парентерального введения и используется прежде всего для профилактики тошноты и рвоты, возникающей на фоне проведения химиотерапии. Ондансетрон редко вызывает побочные реакции.
Одним из новых противорвотных препаратов, применяемых для профилактики рвоты, вызываемой химиотерапевтическими средствами, является трописетрон [2].
1. Нормальная и патологическая физиология1.1 Нормальная физиология
Основное назначение желудочно-кишечного тракта – превращение пищи в такие молекулы, которые могут всасываться в кровь и транспортироваться в другие органы. Начинаются эти процессы с механической обработки пищи (измельчения, перемешивания, перемещения) и секреции пищеварительных соков. Содержащиеся в соках ферменты расщепляют белки, жиры и углеводы на мелкие фрагменты, способные всасываться. Вместе с водой, минеральными солями и витаминами конечные продукты переваривания поступают из просвета кишечника через клетки его слизистой оболочки в кровь и лимфу [3].
Желудочно-кишечный тракт представляет собой сплошную трубку, соединяющую ротовое отверстие с анальным, и состоит из ротовой полости, глотки, пищевода, желудка, тонкого и толстого кишечника.
Одни отделы желудочно-кишечного тракта (ротовая полость и пищевод) служат в основном для транспортировки пищи, другие (желудок и толстый кишечник) – для ее хранения, третьи (тонкий кишечник) – для переваривания и всасывания. Регуляция этих функций осуществляется
1) посредством целого ряда гормонов и биологически активных пептидов,
2) за счет сократительной активности гладкомышечных клеток и
3) вегетативной нервной системой.
Ротовая полость, глотка и пищевод образуют функциональную единицу, назначение которой – предварительная обработка пищи перед прохождением ее по желудочно-кишечному тракту. Здесь пища подвергается измельчению и смачиванию слюной, после чего поступает в желудок.
Жевание. На этом подготовительном этапе пища разрезается на куски и перетирается. Ритмичный процесс жевания осуществляется в основном как непроизвольный рефлекторный акт.
При помощи языка пищевой комок удерживается между челюстями в пределах жевательной поверхности зубов. Благодаря слюноотделению, стимулированному жеванием, пища приобретает консистенцию, необходимую для проглатывания.
Жевание завершается глотанием – переходом пищевого комка из полости рта в желудок [4].
Глотание возникает в результате раздражения чувствительных нервных окончаний тройничного, гортанных и языкоглоточного нервов. По афферентным волокнам этих нервов импульсы поступают в продолговатый мозг, где расположен центр глотания. От него импульсы по эфферентным двигательным волокнам тройничного, языкоглоточного, подъязычного и блуждающего нервов достигают мышц, обеспечивающих глотание.
Перед глотанием глоточно-пищеводный сфинктер закрыт, во время глотания давление в глотке повышается до 45 мм рт. ст., сфинктер открывается, и пищевой комок поступает в начало пищевода, где давление не более 30 мм рт. ст.
Пищеварительными функциями желудка являются депонирование, механическая и химическая обработка пищи и постепенная эвакуация содержимого желудка в кишечник. Пища, находясь несколько часов в желудке, набухает, разжижается, многие ее компоненты растворяются и подвергаются гидролизу ферментами слюны и желудочного сока.
Вся масса пищи в желудке не смешивается с соком. По мере разжижения и химической обработки пищи ее слой, прилегающий к слизистой оболочке, движениями желудка перемещается в антральную часть, откуда пищевое содержимое эвакуируется в кишечник. Таким образом, пищеварение в полости желудка осуществляется некоторое время за счет слюны, но ведущее значение имеет секреторная и моторная деятельность самого желудка.
В проксимальном отделе желудка отсутствует какой-либо ритм возбуждения и перистальтики. В нем поддерживается тонус, зависящий от наполнения желудка. Иными словами, основное назначение проксимального отдела желудка – хранение поступившей в него пищи. Еще до того, как пищевой комок поступает из пищевода в желудок, давление в нем падает (рецептивное расслабление). Благодаря рецепторам растяжения мышечный тонус желудка изменяется таким образом, что его объем увеличивается без какого-либо повышения давления (адаптивное расслабление). При поступлении порции пищи в желудок относительно твердые ее компоненты располагаются слоями, а жидкость и желудочный сок обтекают их снаружи и поступают в дистальный отдел желудка. Медленные тонические сокращения создают постоянное давление, под действием которого пища постепенно перемещается в сторону привратника [3].
Сильные круговые перистальтические волны в дистальном отделе желудка проталкивают его содержимое в сторону привратника и двенадцатиперстной кишки. Жидкость быстро эвакуируется в двенадцатиперстную кишку, и ее объем в желудке уменьшается. Твердые компоненты пищи не проходят через привратник до тех пор, пока не будут измельчены до частиц размером не более 2 – 3 мм. Когда перистальтические волны достигают дистального участка антрального отдела, привратник сокращается. Пилорический отдел, образующий самую узкую часть желудка в месте его соединения с двенадцатиперстной кишкой, закрывается еще до того, как антральный отдел полностью отгораживается от тела желудка. Пища под давлением перемещается обратно в желудок, при этом твердые частицы трутся одна о другую и еще больше измельчаются.
Опорожнение желудка регулируется вегетативной нервной системой, интрамуральными нервными сплетениями и гормонами. Скорость опорожнения желудка зависит от разности давления в желудке и в двенадцатиперстной кишке и от резистентности пилорического отдела.
В регуляции опорожнения желудка помимо его наполнения, размеров частиц и вязкости содержимого играют роль рецепторы тонкого кишечника.
1.2 Патологическая физиология
Нарушение нормальной функции пищеварительного тракта может приводить к различным заболеваниям и клиническим проявлениям: расстройствам пищеварения или всасывания, дискинезии (понос, запор, рвота, недержание кала) и таким явлениям, как изжога, ощущение тяжести и переполнения, колики и тошнота [3].
Рвота – форсированный выброс желудочного содержимого через рот. Позыв к рвоте – усиленная ритмичная одышка, предшествующая рвоте [2].
Рвота контролируется специальными структурами продолговатого мозга: рвотным центром и так называемой хеморецепторной пусковой (триггерной) зоной. Рвотный центр состоит из большого числа эфферентных ядер с множественными взаимосвязями между ними. Когда вся цепь активизируется афферентными стимулами, возникает акт рвоты. Но некоторые пусковые факторы стимулируют только отдельные звенья нервной цепи, приводящей к возникновению рвоты, в результате возникает или тошнота, или слюноотделение. К рвотному центру стимулы подходят по трем афферентным путям:
1) по вагусным и симпатическим афферентным волокнам от желудочно-кишечного тракта, вестибулярного аппарата и сердца;
2) по расположенным в самой задней части дна четвертого желудочка хеморецепторам триггерной зоны, которые могут стимулироваться эметогенными токсинами или лекарственными препаратами (препараты наперстянки, химиотерапевтические средства);
3) по участкам, расположенным высоко в центральной нервной системе.
Два центра в ретикулярной формации продолговатого мозга служат хеморецептивными триггерными зонами (реагируют при уремии, лекарственной терапии и ионизирующем излучении) и одновременно интегрирующим центром. Афферентная иннервация исходит практически изо всех точек организма. Очень важны волокна n. vagi, но после ваготомии рвота не прекращается. Симпатическая афферентная иннервация – посредник в проведении импульсации, заканчивающейся актом рвоты, связанной с растяжением кишечника. Рвота возникает, когда нервные импульсы одновременно по соматическим и висцеральным эфферентным путям вызывают закрытие голосовой щели, поднятие диафрагмы, спазм пилорического сфинктера и расслабление желудка, вслед за чем перистальтическая волна движется от средней части желудка к incisura cardiaca ventriculi, в сочетании с сокращением брюшной, диафрагмальной и межреберной мускулатур. Одновременно со рвотой отмечаются признаки активации вегетативной нервной системы [5].
Патофизиология рвоты: желудочное содержимое проталкивается в пищевод, когда вслед за расслаблением дна желудка и желудочно-пищеводного сфинктера быстро повышается внутрибрюшное давление из-за сокращения брюшных и диафрагмальных мышц. Увеличение внутригрудного давления ведет к дальнейшему продвижению пищевой массы в полость рта. Рефлекторный подъем мягкого неба и закрытие голосовой щели защищает носоглотку и трахею и завершает акт рвоты [2].
Рвота характеризуется наличием тошноты и автономных симптомов, таких как слюноотделение, и сопровождается выраженными сокращениями брюшных и грудных мышц, связанными с возникновением позывов на рвоту.
Тошнота возникает в том случае, когда мозг хочет довести до сознания человека, что в организм попали какие-либо токсичные вещества, как в случае пищевого отравления. Этим же способом мозг предупреждает человека, что он делает нечто, таящее в себе угрозу для здоровья. Если тошнота возникает как реакция на сдачу экзаменов, то это можно расценить как сигнал о том, что в крови слишком высок уровень адреналина. Предупреждение верное, но излишнее. Тошнота, возникающая в результате быстрого движения, например во время езды на автомобиле, также относится к категории излишних предупреждений. Таким образом, тошнота – это самый распространенный способ, которым мозг подает человеку сигнал о первых проявлениях какого-либо отклонения от нормы. Ощущения тошноты могут служить признаком заболевания любого органа человеческого организма, как, например, почек, печени, кишечника или даже сердца. Если тошнота носит упорный характер, это может служить признаком рака. Закупорка, возникающая в результате сужения сосудов, воспаление или опухоль в любом месте желудочно-кишечного тракта могут привести к рвоте и быть ее причиной.
Еще одна из наиболее часто встречающихся причин тошноты – это пониженное содержание сахара в крови. Низкий уровень сахара, часто связанный с чувством голода, может вызвать тошноту. Это выглядит парадоксально, если учесть, что тошнота обычно приводит к понижению аппетита. Скорее всего, тошнота служит более агрессивным по сравнению с обычным ощущением голода способом мозга привлечь внимание человека к факту, что ему необходимо принять пищу. Головной мозг прибегает к этому способу, когда другие сигналы остались без ответа. Чувство подступающей тошноты не всегда имеет отношение к проблемам пищеварения, однако очищение желудка помогает в любом случае.
Фактически любое токсическое вещество в организме, каким бы ни было его происхождение, вызывает тошноту и может привести к рвоте. Токсины могут появиться в результате нарушений функции почек или печени, излишнего употребления алкоголя или как реакция на бактерии, попавшие в организм вместе с пищей, как в случае пищевого отравления.
Как правило, рвота свидетельствует об интоксикации организма и является способом выведения из него отравляющих веществ. Рвота часто сопровождается усиленным выделением пота, что является еще одним способом выведения токсинов. При этом также возникают позывы к мочеиспусканию и дефекации. Если токсины находятся в желудке или верхней части кишечника, они действительно выводятся с рвотными массами, что приводит к улучшению состояния. Однако очень часто токсины попадают в кровь, что приводит к возбуждению рвотных центров головного мозга. В этом случае рвота не дает положительного эффекта.
К заболеваниям, которые обычно сопровождаются хронической тошнотой и рвотой, относятся:
1. функциональные нарушения эвакуации желудочного содержимого (парез желудка);
2. нарушение двигательной активности тонкой кишки;
3. психогенная рвота. Главным анамнестическим признаком у таких пациентов является длительно существующая повторяющаяся рвота после еды.
1) Парез желудка представляет собой нарушение эвакуации желудочного содержимого при отсутствии механического препятствия в желудке или в тонкой кишке. Парез желудка может развиваться при приеме некоторых лекарств (в частности наркотиков), после резекции желудка (в таких случаях может развиваться застой в культе желудка), у пациентов с сахарным диабетом, склеродермией, амилоидозом или при отсутствии каких-либо явных причин (идиопатический парез желудка). Часто бывает трудно отличить идиопатический парез желудка от психогенной рвоты. В постановке диагноза помогает изучение скорости опорожнения желудка с твердыми продуктами питания. Чаще всего для этой цели используется сэндвич с жареным яйцом, меченным радиоактивным изотопом.
2) Парез тонкой кишки (псевдообструкция), как следует из названия, развивается при отсутствии каких-либо механических препятствий в тонкой кишке. Отсутствие механического препятствия необходимо подтвердить с помощью разнообразных диагностических исследований (в т. ч. тонкой кишки с рентгеноконтрастным веществом), выполняемых в период реконвалесценции. У пациентов со стойким парезом тонкой кишки могут наблюдаться вздутие живота, боли в животе, нарушение стула, ортостатическая гипотензия или симптомы со стороны мочевого пузыря (дизурические расстройства). Парез тонкой кишки может развиваться у пациентов на фоне приема некоторых медикаментозных препаратов и при различных системных заболеваниях, например склеродермии, сахарном диабете и амилоидозе. При проведении дифференциальной диагностики у пациентов с парезом тонкой кишки необходимо также исключить дивертикулез тощей кишки. При исключении всех вышеперечисленных заболеваний причиной пареза тонкой кишки может быть миопатия или нейропатия тонкой кишки. В таких случаях поставить диагноз позволяет выявление нарушений двигательной активности пищевода. Если двигательная активность пищевода нормальная, а хирург имеет основания предполагать наличие мио- или нейропатии тонкой кишки, необходимо исследовать двигательную активность тонкой кишки, а также (иногда) выполнить гистологическое исследование участка стенки тонкой кишки, взятого во время операции.
3) Психогенная рвота нередко бывает причиной повторяющейся рвоты, особенно у молодых женщин. В основе ее, как правило, лежат различные эмоциональные нарушения. Иногда психогенная рвота бывает проявлением глубокой депрессии или истерических реакций. В постановке диагноза психогенной рвоты большое значение играет сбор анамнеза, а также факт наличия у пациента длительного эмоционального напряжения на момент возникновения рвоты. Кроме того, рвота чаще всего появляется сразу после приема пищи, и при необходимости ее можно легко подавить. И наконец, психогенная рвота на удивление мало тревожит самого пациента и в основном беспокоит членов его семьи. Одним из основных сопутствующих симптомов психогенной рвоты являются боли в животе. Очень важно вовремя поставить правильный диагноз, с тем, чтобы пациент избежал различных инвазивных диагностических манипуляций и операций, которые способны ухудшит состояние пациента и привести к возникновению различных осложнений.
Рвота может быть причиной заболеваний органов брюшной полости: гастритом, язвенной болезнью, диафрагмальной грыжей, холециститом, острыми хирургическими заболеваниями (аппендицит, непроходимость кишечника, желчные и почечные колики), пилороспазмом, пилоростенозом; инфекционными заболеваниями: дизентерией, скарлатиной, коклюшем, гриппом и т. д.; заболеваниями нервной системы: менингитом, энцефалитом, опухолями; с функциональными нарушениями нервной системы: сотрясением мозга, невропатиями, мигренью, «кинетозами» (рвота появляется в поездке на транспорте); заболеваниями с нарушением обмена веществ (ацетонемическая рвота, сахарный диабет, уремия), а также при недостаточности кровообращения, отравлениях. Как самостоятельная форма болезни выделяется ацетонемическая рвота [6].
Рвота ацетонемическая тяжелая рвота, выделяется как синдром, который нередко повторяется с определенной периодичностью, сопровождается симптоматической ацетонемией. Представляет болезнь детей дошкольного и школьного возраста, чаще наблюдается у детей-невропатов в возрасте 2 – 10 лет.
Патогенез до сих пор не ясен. Предрасполагающими факторами могут быть эмоциональные шоки, физическая и умственная усталость, неправильное питание (избыточное употребление жиров, недостаточное – углеводов и др.). Предполагают, что в промежуточном мозге возникают доминантные очаги возбуждения. С появлением неукротимой рвоты возникают глубокие нарушения промежуточного обмена, ацетонемия, ацетонурия.
Последствия рвоты. Метаболические. При выраженной (сильной и повторной) рвоте развиваются гиповолемия, гипокалиемия, метаболический алкалоз и уменьшается содержание общего натрия в организме. Калий выводится из организма почками в обмен на натрий, если в организме содержится недостаточно иона Н+ для обмена. Дальнейшее уменьшение объема жидкости в организме может привести к парадоксальной ацидурии при попытке сохранить в организме натрий. Другие. Повторные интенсивные приступы рвоты могут привести к разрывам слизистой оболочки желудка типа Мэллори–Вейсс (Mallory–Weiss) или разрыву дистального отдела пищевода синдром Бурхаве (Boerhaave).
2. История ОТКРЫТИЯ РВОТНЫХ И ПРОТИВОРВОТНЫХ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ50-е годы – начало психофармакологии. В 20-х годах ХХ столетия великий русский физиолог И.П. Павлов сделал выдающееся открытие. Применив в качестве средства, стимулирующего центральную нервную систему кофеин и успокаивающее средство натрия бромид, он показал возможность «управления» функциями центральной нервной системы. Это открытие положило начало современной психофармакологии [7].
В начале 50-х годов появился первый высокоэффективный антипсихотический препарат – хлорпромазин (аминазин).
Аминазин впервые синтезирован во Франции; сообщение о нем появилось в печати в 1952 году [8].
История хлорпромазина начинается с конца 30-х годов, когда искали противогистаминные препараты среди производных фенотиазина. Тогда были созданы противогистаминные препараты этизин и прометазин [7].
При изучении прометазина (фенергана) было установлено, что он не только обладает противогистаминным действием, но и успокаивает нервную систему, оказывает адреноблокирующее действие. Французский физиолог Лабори (Laborit) предложил в связи с этим применять фенерган для «нейроплегии» и управляемой гипотензии с целью облегчения проведения хирургических операций. В развитие работ по аналогам фенергана было синтезировано большое количество других производных фенотизиана и среди них соединение R.P. 4560. При изучении этого соединения фармаколог Курвуазье (Courvoisier) обнаружила, что, проявляя относительно слабую противогистаминную активность, оно оказывает выраженное успокаивающее действие на центральную нервную систему, кроме того, обладает адренолитической, гипотермической и другими видами активности. Соединение получило в связи с этим название ларгактил («широко действующее»). В дальнейшем в соответствии с химической структурой оно получило международное название хлорпромазин (в России – аминазин).
Хлорпромазин (аминазин) остается до сих пор одним из основных антипсихотических препаратов, хотя вслед за ним была создана целая «гамма» других фенотиазиновых препаратов, оказывающих антипсихотическое действие.
В настоящее время из производных этой группы применение имеют: хлорпромазин (аминазин), пропазин, левомепромазин (тизерцин), алимемазин (терален), этаперазин (перфеназин), трифтазин (стелазин), пипотиазин (пипортил), фторфеназин (модитен), тиоперазин (мажептил) и ряд других. Различаясь несколько по структуре, они обладают также несколько разным спектром действия, различной длительностью действия, по-разному переносятся больными.
Дипразин синтезирован в 1944 г. во Франции одновременно с динезином [8].
По современной классификации эти препараты относятся к группе «нейролептики». К ней же относятся ряд созданных в дальнейшем антипсихотических препаратов других химических групп: производные бутирофенона (галоперидол, трифлуперидол, дроперидол и др.); производные дифенилбутилпиперидина (флушпирилен, пимозид и др.); производные бензамида (сульпирид, тиаприд) [7].
В настоящее время изучен в основном нейрохимический механизм действия нейролептиков. Связан он главным образом с блокадой центральных дофаминовых (Д2) рецепторов, частично также – с блокадой адренергических рецепторов.
Первое сообщение о синтезе дофамина было сделано одновременно и независимо друг от друга G.Barger, P.C.Ewins (1910) и E.Mannich, W.Jacobsohn (1910) [9].
В 1984 году был синтезорован агонист Д2-рецепторов бромокриптин (Horn A.S., Tepper R., Kebabian J.W. et al.). В 1989 году синтезирован антагонист Д2-рецепторов: домперидон (Teral M., Hidaka K., Nakamura Y.) и в 1990 году – сульпирид (Amalric M., Merhow M., Polis J. et al.).
Гистамин относится к наиболее старым биогенным аминам, история изучения которого начинается с 1907 года, когда Виндаус и Фогт синтезировали его из имдазолиронионовой кислоты. Первая работа, посвященная изучению биологической активности гистамина появилась в 1910 году (Dale H. et al.). Только через 26 лет после этого был синтезирован антагонист гистамина, который способствовал идентификации гистаминового рецептора (D.Bovet, A.Staub; 1936) [9].
Антигистаминные препараты были открыты в конце 1930-х годов. К 1950-м годам были предложены высокоэффективные антагонисты гистамина – трипеленнамин и дифенилгидрамин, которые иницирировали широкие исследования в области синтеза этого типа препаратов [10].
Крупным фундаментальным достижением 50-х годов, сыгравшим большую роль в прогрессе нейрофармакологии, явилось открытие медиаторной роли серотонина [7].
В конце XIX века физиологи обнаружили, что при образовании кровяных сгустков из них выделяется вещество, вызывающее сужение кровеносных сосудов, и дали ему название вазотонин. В 40-х годах ХХ столетия было высказано предположение, что это вещество играет роль в патогенезе артериальной гипертензии и началось его подробное изучение. В 1948 году оно было выделено в кристаллическом виде (Раппорт и др.) и названо серотонином. При химическом изучении было установлено, что серотонин является производным индола, именно 5-гидрокситриптамином (5-НТ). Вскоре серотонин был обнаружен в тканях мозга (Тварог, Пэйдж) и было высказано предположение, что он участвует в деятельности центральной нервной системы в качестве нейромедиатора. Синтез серотонина был осуществлен в 1951 году.
Исследование физиологической и фармакологической роли серотонина расширило представления о химической природе передачи нервного возбуждения, об эндогенных физиологически активных веществах, появились новые возможности для объяснения механизмов действия некоторых лекарственных средств и для создания новых лекарственных препаратов.
При изучении эндогенных серотонинергических процессов было выявлено три вида серотониновых рецепторов: С1(5-НТ1)-, С2(5-НТ2)- и С3(5-НТ3)-рецепторов, локализующихся как в периферических органах, так и в центральной нервной системе.
Высокая концентрация 5-НТ1- и 5-НТ3-рецепторов обнаружена в гладкой мускулатуре и слизистой оболочке желудочно-кишечного тракта; 5-НТ2-рецепторов – в гладких мышцах стенок кровеносных сосудов, в бронхах и тромбоцитах; 5-НТ3-рецепторов – в периферических тканях и в центральной нервной системе.
Прикладными фармакологическими результатами открытия и изучения серотонина явилось применения самого серотонина и его аналогов в качестве лекарственных средств и создание разных эффективных антисеротониновых препаратов (ондансетрон, трописетрон и др.)
Большой интерес вызвали синтезированные в 1989 году блокаторы серотониновых 5-НТ3-рецепторов препараты ондансетрон (латран), трописетрон, гранисетрон и др. Через эти рецепторы, плотно локализованные в триггерных зонах рвотного центра мозга, реализуются тошнота и рвота, обусловленные поступлением в организм химических соединений, особенно противоопухолевых препаратов. Ондансетрон, трописетрон и их аналоги нашли широкое применение для профилактики и терапии этих осложнений при химио- и лучевой терапии онкологических заболеваний.
В целом открытие и изучение серотонина внесли крупный вклад в прогресс фармакологии.
Начатое в 50-х годах изучение серотонинергической системы продолжается до сих пор [9].
Открытие алкалоидов в начале ХIХ столетии имело большое значение для развития химии, так как ранее считали, что растительные вещества в отличие от веществ животного происхождении не содержат азота [8]. Первым таким веществом, выделенным в 1804 г. Сертюрнером в виде смеси кристаллических веществ из опия, был морфин.
В 1817 году Сертюрнер получил более чистый продукт и назвал его морфином ввиду обнаруженных снотворных свойств.
Апоморфин получен впервые в 1869 г. дегидратацией морфина.
Эмпирическая формула морфина была установлена в 1848 г., но в течении ряда десятилетий, несмотря на многочисленные работы химиков, не удавалось расшифровать его строение. Структура морфина была установлена лишь в 1925 – 1927 гг.
Синтез морфина осуществлен Гетисом в 1950 г.
Скополамин выделен Э. Шмидтом в 1888 г. из корней Scopolia atropoides.
Таблица 2.1
Хронологическая таблица истории открытия рвотных и противорвотных лекарственных препаратов
| Событие | Кем было открыто | Дата открытия |
| Выделение морфина в виде смеси кристаллических веществ из опия | Сертюрнер | 1804 |
| Был получен более чистый морфин | Сертюрнер | 1817 |
|
Установлена эмпирическая формула морфина |
1848 | |
| Впервые получен апоморфин дегидратацией морфина | 1869 | |
| Скополамин выделен из корней Scopolia atropoides | Э. Шмидт | 1888 |
| Установлена структура морфина | 1925 1927 гг. | |
| Осуществлен в синтез морфина | Гетис | 1950 |
| Открытие блокаторов гистаминовых H1-рецепторов | 30-е годы | |
| Синтезирован аминазин | Начало 1950-х годов | |
| Сообщение об аминазине впервые появилось в печати | 1952 | |
| Синтезирован дипразин | 1944 | |
| Открытие антигистаминных препаратов | Конец 1930-х годов | |
| Синтезирован галоперидол | 1950-е годы | |
| Синтезирован дифенилгидрамин | 1950-е годы | |
| Синтезирован метоклопрамид | 1960-е годы | |
| Синтезирован бромокриптин | Horn A.S., Tepper R., Kebabian J.W. et al. | 1984 |
| Синтезирован домперидон | Teral M., Hidaka K., Nakamura Y. | 1989 |
| Синтезированы ондансетрон, трописетрон, гранисетрон | 1989 | |
| Синтезирован сульпирид | Amalric M., Merhow M., Polis J. et al. | 1990 |
Существует две классификации рвотных и противорвотных лекарственных средств.
3.1 Классификация препаратов по Машковскому
1. Лекарственные средства, действующие преимущественно на центральную нервную систему [11].
1.1 Психотропные препараты
А. Нейролептические средства
а) производные фенотиазина
| Аминазин | Пропазин |
| Левомепромазин | Алимемазин |
| Этаперазин | Френолон |
| Трифтазин | Фторфеназин |
| Фторфеназин-деканоат | Тиопроперазин |
| Перициазин |
б) приозводные гиоксантена
Хлорпротиксен
в) производные бутирофенона и дифенилбутилпиперидина
Галоперидол
Трифлуперидол
Дроперидол
Бенперидол
г) нейролептики разных химических групп
Клозапин
Сульпирид
Б. Седативные средства
| Натрия бромид | Бромкамфора |
| Корневища с корнями валерианы | Валокормид |
| Валоседан | Корвалол |
| Трава пустырника | Трава пассифлоры |
1.2 Рвотные и противорвотные препараты
Апоморфин
Тиэтилперазин
Метоклопрамид
2. Лекарственные средства, действующие преимущественно на периферические нейромедиаторные процессы
2.1 Средства, действующие на периферические холинергические процессы
В. Антихолинергические средства, блокирующие преимущественно периферические холинореактивные системы
а) Алкалоиды группы атропина и содержащие их растения
Скополамин
2.2 Средства, действующие преимущественно на периферические адренергические процессы
А. Адреналин и адреномиметические вещества
Дименгидринат
2.3 Гистамин и антигистаминные препараты
Димедрол
Дипразин
3. Лекарственные средства, действующие преимущественно в области чувствительных нервных окончаний
3.1 Местноанестезирующие
Анестезин
3.2 Средства, действие которых связано преимущественно с раздражением нервных окончаний слизистых оболочек и кожи
А. Средства, содержащие эфирные масла
Ментол
Валидол
4. Противомикробные, противопаразитарные и противовирусные средства
4.1 Антисептические средства
| А. Препараты меди | Б. Препараты цинка |
| Меди сульфат | Цинка сульфат |
4.2 Классификация по действию на нейромедиаторные процессы
1. Препараты, блокирующие серотониновые рецепторы [12].
1.1 Трописетрон
1.2 Ондансетрон
1.3 Гранисетрон
2. Препараты, блокирующие дофаминовые рецепторы
| 2.1 Диметпрамид | 2.2 Домперидон |
| 2.3 Метоклопрамид | 2.4 Сульпирид |
| 2.5 Тиэтилперазин | 2.6 Бромокриптин |
| 2.7 Апоморфин | 2.8 Галоперидол |
3.Препараты, блокаторующие гистаминовые Н1-рецепторы
3.1 Димедрол
3.2 Дипразин
3.3 Прометазин
4. М-холиноблокаторы
4.1 Скополамин
4. Механизм биологической активности РВОТНЫХ И ПРОТИВОРВОТНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ4.1 Лекарственные средства, блокирующие серотониновые рецепторы
Препараты данной группы (ондансетрон, гранисетрон, трописетрон) являются конкурентные антагонисты серотониновых 5НТ3-рецепторов в периферических тканях и ЦНС и устраняют рвоту, индуцируемую химиотерапией, а также синдром желудочной диспепсии в послеоперационном периоде [12].
4.1.1 Трописетрон (навобан).
Механизм действия связан с избирательным блокированием периферических и центральных серотониновых рецепторов [13]. Трописетрон – сильнодействующий и высокоселективный конкурентный антагонист 5-HT3-рецепторов – подкласса рецепторов к серотонину, расположенных на периферических нейронах и в ЦНС. Хирургические вмешательства и лечение с применением определенных препаратов, в том числе некоторых химиотерапевтических средств, могут способствовать выделению серотонина (5-HT) из энтерохромаффиноподобных клеток, расположенных в слизистой оболочке желудочно-кишечного тракта. Это инициирует рвотный рефлекс и сопутствующее ему ощущение тошноты. Трописетрон селективно блокирует возбуждение пресинаптических 5-HT3-рецепторов на периферических нейронах, принимающих участие в возникновении этого рефлекса, а также может оказывать дополнительное прямое действие на 5-HT3-рецепторы, расположенные в ЦНС и опосредующие влияние блуждающего нерва на area postrema. Как считается, эти влияния лежат в основе механизма противорвотного действия трописетрона. Длительность действия составляет 24 часа, что позволяет применять его один раз в день.
Фармакокинетика. Навобан всасывается из желудочно-кишечного тракта почти полностью (более, чем на 95%). Период полуабсорбции составляет около 20 мин.
Неспецифическое связывание трописетрона с белками плазмы (преимущественно с альфа1–гликопротеинами) составляет 71%. Объем распределения у взрослых составляет от 400 до 600 л; у детей в возрасте от 3 до 6 лет – около 145 л, у детей в возрасте от 7 до 15 лет – примерно 265 л. Максимальная концентрация в плазме достигается в пределах 3 часов. Биодоступность зависит от величины дозы: после приема препарата в дозе 5 мг она достигает приблизительно 60% и повышается (вплоть до 100%) после приема препарата в дозе 45 мг. Значения биодоступности и конечного периода полувыведения у детей сходны с соответствующими показателями, наблюдавшимися у здоровых добровольцев.
Метаболизм трописетрона осуществляется путем гидроксилирования в 5, 6 или 7 положениях индольного кольца, с последующей реакции конъюгации с образованием глюкуронида или сульфата и выведением с мочой или с желчью (соотношение содержания метаболитов в моче и кале составляет 5:1). Активность метаболитов трописетрона в отношении 5-HT3-рецепторов значительно снижена, и они не участвуют в реализации фармакологического действия препарата.
При повторных назначениях навобана в дозах, превышающих 10 мг два раза в день, может произойти насыщение ферментной системы печени, участвующей в метаболизме трописетрона, что может привести к дозозависимому повышению уровней трописетрона в плазме. Однако, даже у лиц, плохо метаболизирующих трописетрон, применение таких доз препарата не приводило к увеличению концентрации препарата в плазме выше переносимых значений. Поэтому полагают, что в том случае, когда для предупреждения возникновения тошноты и рвоты во время противоопухолевой химиотерапии на протяжении 6 дней будет применяться рекомендуемая доза препарата, составляющая 5 мг один раз в день, накопление трописетрона не будет иметь клинического значения.
У лиц, быстро метаболизирующих трописетрон, период полувыведения (бета-фаза) составляет около 8 часов; у пациентов, плохо метаболизирующих трописетрон, величина этого показателя может удлиняться до 45 ч.
4.1.2 Ондансетрон (эметрон, зофран).
Оказывает сильное противорвотное действие, механизм которого окончательно не установлен. Препараты, применяемые для химиотерапии, и радиологическое воздействие могут вызывать высвобождение серотонина в тонкой кишке, запуская тем самым рвотный рефлекс через активацию серотониновых 5НТ3-рецепторов и возбуждение афферентных окончаний блуждающего нерва. Ондансетрон блокирует пусковые механизмы этого рефлекса. Активация афферентных окончаний блуждающего нерва, в свою очередь, может вызвать выброс серотонина в зоне пострема, находящейся на дне IV желудочка, и, следовательно, запустить рвотный рефлекс через центральный механизм. Подавление тошноты и рвоты, спровоцированные цитотоксической химиотерапией и радиотерапией, по-видимому, осуществляется благодаря антагонистическому действию ондансетрона на серотониновые 5НТ3-рецепторы нейронов центральной и периферической нервной системы. При психомоторном тестировании показано, что ондансетрон не ухудшает работоспособность и не оказывает седативного действия. Препарат не влияет на концентрацию пролактина в плазме крови.
Фармакокинетика. После приема препарата внутрь Сmax достигается примерно через 1,5 ч. Абсолютная биодоступность составляет около 60 %. После ректального введения 1 суппозитория ондансетрон определяется в плазме через 15-60 мин. Концентрация активного вещества увеличивается линейно, Сmax достигается примерно через 6 ч. и составляет 20-30 нг/мл. Снижение концентрации в плазме происходит с меньшей скоростью, чем после приема препарата внутрь, вследствие продолжающегося всасывания ондансетрона. Абсолютная биодоступность ондансетрона при ректальном введении составляет приблизительно 60% и не зависит от пола. После в/м введения Сmax достигается через 10 мин. Vd как после приема внутрь, так и после парентерального введения составляет 140 л. Связывание с белками плазмы – 70–76 %. Биотрансформируется в печени. Как после приема внутрь, так и после парентерального введения T1/2 составляет 3 ч. После ректального введения T1/2 определяется скоростью всасывания ондансетрона, а не системным клиренсом, и составляет приблизительно 6 ч. В неизмененном виде с мочой выводится менее 5 % от введенной дозы. Фармакокинетические параметры ондансетрона не изменяются при его многократном введении.
4.2 Лекарственные средства, блокирующие дофаминовые рецепторы
Препараты этой группы (диметпрамид, домперидон, метоклопрамид, сульпирид, тиэтилперазин, бромокриптин, апоморфин) блокируют дофаминовые рецепторы в триггерной зоне рвотного центра. [12].
4.2.1 Диметпрамид.
По структуре и механизму действия близок к сульпириду и метоклопрамиду [13]. Оказывает противорвотное действие и применяется для предупреждения и купирования тошноты и рвоты в послеоперационном периоде, при лучевом лечении и химиотерапии онкологических больных и др.
4.2.2 Домперидон (мотилиум).
Оказывает противорвотное действие, успокаивает икоту и устраняет в некоторых случаях тошноту. Оказывает регулирующее и нормализующее влияние на двигательную активность желудочно-кишечного тракта, что связано с блокирующим влиянием на дофаминовые рецепторы (Д2) желудочно-кишечного тракта. По действию близок к метоклопрамиду. В отличие от метоклопрамида не проникает через гематоэнцефалический барьер и не вызывает экстрапирамидных расстройств.
4.2.3 Метоклопрамид.
Противорвотное средство, способствует уменьшению тошноты, икоты; стимулирует перистальтику ЖКТ. Противорвотное действие обусловлено блокадой дофаминовых рецепторов и повышением порога хеморецепторов триггерной зоны. Полагают, что метоклопрамид ингибирует расслабление гладкой мускулатуры желудка, вызываемое дофамином, усиливая таким образом холинергические реакции гладкой мускулатуры ЖКТ. Способствует ускорению опорожнения желудка путем предотвращения расслабления тела желудка и повышения активности антрального отдела желудка и верхних отделов тонкой кишки. Уменьшает рефлюкс содержимого в пищевод за счет увеличения давления сфинктера пищевода в состоянии покоя и повышает клиренс кислоты из пищевода благодаря увеличению амплитуды его перистальтических сокращений. Метоклопрамид стимулирует секрецию пролактина и вызывает повышение уровня циркулирующего альдостерона, что может сопровождаться кратковременной задержкой жидкости.
По химической структуре близок к сульпириду и диметпрамиду.
Фармакокинетика. После приема внутрь быстро всасывается из ЖКТ [12]. Связывание с белками плазмы около 30 %. Cmax его в крови определяется через 30–120 минут после приема. Препарат метаболизируется в печени, в виде метаболитов и в неизмененном виде (менее 20 %) выводится почками. Т1/2 равен 2–4 часа. Ускорение эвакуации желудочного содержимого после введения метоклопрамида длится 3 часа, противорвотный эффект около 12 часов.
4.2.4 Сульпирид.
Является производным сульфонилбензамида. По строению и некоторым фармакологическим свойствам сульпирид близок также к метоклопрамиду. Подобно метоклопрамиду оказывает противорвотное действие; кроме того, он обладает фармакологическими свойствами, характерными для психотропных препаратов; оказывает умеренное антисеротониновое и каталептогенное действие, несколько ослабляет стимулирующие эффекты фенамина. Седативного действия не оказывает, не усиливает влияния барбитуратов и аналгетиков. Противосудорожной активностью не обладает.
Клинически сульпирид характеризуется как препарат с регулирующим влиянием на ЦНС, у которого умеренная нейролептическая активность сочетается с некоторыми антидепрессивными и стимулирующими свойствами. Нейролептический эффект, возможно, объясняется тем, что препарат является антагонистом дофаминовых рецепторов.
Холиномиметические эффекты метоклопрамида и сульпирида ограничены проксимальным отделом кишечника и устраняются м-холиноблокаторами.
4.2.5 Тиэтилперазин (торекан, горестен, трестен).
Относится к производным фенотиазина. Не обладает выраженной седативной активностью, лишь слабо потенцирует действие снотворных и аналгетических веществ, не оказывает выраженного каталептогенного действия и при клиническом применении обычно не вызывает сильных экстрапирамидных нарушений. Вместе с тем тиэтилперазин оказывает сильное противорвотное действие; по этому показателю он значительно более активен, чем аминазин, и превосходит метеразин. Препарат эффективен при рвоте различного происхождения. В условиях эксперимента он подавляет рвоту, вызванную возбуждением рвотного центра (апоморфином) и раздражением рецепторов желудочно-кишечного тракта (сульфатом меди). Механизм противорвотного действия тиэтилперазина складывается из успокаивающего влияния на рвотный центр и одновременного действия на хеморецепторную пусковую (триггерную) зону продолговатого мозга.
4.2.6 Бромокриптин (парлодел).
Полусинтетическое производное алкалоида спорыньи – эргокриптина. Является специфическим агонистом дофаминовых рецепторов (главным образом типа Д2). Препарат активно влияет на кругооборот дофамина и норадреналина в ЦНС, уменьшает выделение серотонина. Стимулирует дофаминовые рецепторы в мозге; подавляет секрецию пролактина и в меньшей степени гормона роста аденогипофизом.
В связи со стимулирующим действием на дофаминовые рецепторы гипоталамуса бромокриптин оказывает характерное тормозящее влияние на секрецию гормонов передней доли гипофиза, особенно пролактина и соматотропина. Эндогенный дофамин является физиологическим ингибитором секреции этих гормонов.
Синтеза пролактина бромокриптин не нарушает. Тормозящее влияние на секрецию гормона снимается блокаторами дофаминовых рецепторов (например, аминазином).
Бромокриптин (как и апоморфин, являющийся стимулятором Д2-рецепторов) оказывает рвотное действие, снижает температуру тела, уменьшает акинезию, вызванную резерпином, тетрабеназином, фенотиазиновыми нейролептиками [13]. Препарат оказывает гипотензивное действие, связанное с влиянием на ЦНС, воздействует на симпатические нервные окончания и гладкую мускулатуру сосудов. Снижает содержание в крови катехоламинов.
В отличие от эргометрина, метилэргометрина и других аналогичных препаратов спорыньи, он не оказывает "маточного" (окситоцического) действия [12]. Наоборот, он угнетает сокращения матки, вызванные метилэргометрином.
Фармакокинетика. После приема препарата внутрь степень абсорбции бромокриптина из ЖКТ составляет 30 %. Концентрация бромокриптина в плазме достигает 4–6 нгэкв/мл, в эритроцитах 2–3 нгэкв/мл. Бромокриптин на 90–96 % связывается с альбумином плазмы крови. Биодоступность бромокриптина составляет 6 % из-за выраженного эффекта "первого прохождения" через печень. Выводится преимущественно с калом (85,6 %) и в незначительной степени – с мочой (2,5–5,5 %).
4.2.7 Апоморфин.
Сохраняет некоторые фармакологические свойства морфина [13]. Он обладает слабой анальгезируюшей активностью, оказывает угнетающее влияние на дыхательный центр. Особенно выражено влияние апоморфина на хеморецепторную пусковую зону продолговатого мозга, возбуждение которой обуславливает его сильное рвотное действие. Непосредственно рвотный центр апоморфин, подобно морфину, угнетает. Если первая доза апоморфина рвотного действия не оказала, то повторное введение бывает неэффективным. Применение апоморфина не дает также эффекта, если подавлена возбудимость рвотного центра (например, при глубоком наркозе) или хеморецепторной пусковой зоны (например, под влиянием нейролептических веществ). В последние годы привлекла к себе внимание способность апоморфина стимулировать дофаминергические структуры мозга. В настоящее время установлено, что апоморфин является специфическим агонистом для Д-рецепторов. Целый ряд поведенческих реакций, наблюдаемых у экспериментальных животных при введении апоморфина (стереотипия, агрессивность и др.), обьясняют его стимулирующим влиянием на дофаминовые рецепторы. С влиянием на дофаминовые рецепторы в определенной степени связано и рвотное действие апоморфина. В связи со способностью проникать через гематоэнцефалический барьер и оказывать центральное дофаминергическое действие делались попытки применять апоморфин для лечения паркинсонизма.
Апоморфин дает антипаркинсонический лечебный эффект, но не вошел для этой цели в медицинскую практику вследствие рвотного действия, нефротоксичности и других побочных явлений. Апоморфином широко пользуются также для экспериментальных целей при фармакологическом изучении и поиске новых, более эффективных психотропных препаратов.
Не исключено, что способность апоморфина взаимодействовать с дофаминовыми рецепторами связана со сходством части его молекулы со структурой дофамина.
4.2.8 Галоперидол.
Один из наиболее активных современных нейролептиков из группы производных бутирофенона. Оказывает выраженное антипсихотическое действие, обусловленное блокадой деполяризации или уменьшением степени возбуждения дофаминовых нейронов (снижение высвобождения) и блокадой постсинаптических D2-рецепторов. Оказывает умеренное седативное действие, связанное с конкурентной блокадой постсинаптических дофаминовых D2-рецепторов в лимбических дофаминергических структурах коры головного мозга и усилением метаболизма дофамина в головном мозге. Оказывает сильное противорвотное действие.
Блокада дофаминовых рецепторов в допаминовых путях черно-полосатой субстанции способствует развитию экстрапирамидных двигательных реакций; блокада дофаминовых рецепторов вызывает уменьшение высвобождения СТГ и увеличение высвобождения пролактина гипофизом. Отмечаются также некоторые проявления блокады a-адренорецепторов вегетативной нервной системы.
Фармакокинетика. При приеме внутрь всасывается из ЖКТ на 60 %. Cmax в плазме при приеме внутрь достигается через 3–6 ч, при в/м введении – через 10–20 мин, при в/м введении галоперидола деканоата – 3–9 дней. Связывание с белками составляет 92 %. Vd при равновесной концентрации – 18 л/кг. Активно метаболизируется в печени. T1/2 при приеме внутрь – 24 ч, при в/м введении – 21 ч, при в/в введении – 14 ч. Галоперидола деканоат выводится в течение 3 нед. Выводится почками – 40 % и с желчью через кишечник – 15 %.
4.3 Препараты, блокаторующие гистаминовые Н1-рецепторы
4.3.1 Дифенгидрамин (димедрол).
Является одним из основных представителей группы противогистаминных препаратов, блокирующих Н1-рецепторы [11]. Он обладает весьма выраженной противогистаминной активностью. Кроме того, он оказывает местноанестезирующее действие, расслабляет гладкую мускулатуру в результате непосредственного спазмолитического действия, блокирует в умеренной степени холинорецепторы вегетативных нервных узлов.
Димедрол хорошо всасывается при приеме внутрь. Проникает через гематоэнцефалический барьер.
Важной особенностью димедрола является его седативное действие, имеющее некоторое сходство с действием нейролептических веществ; в соответствующих дозах он оказывает снотворный эффект. Является также умеренным противорвотным средством. В действии димедрола на нервную систему существенное значение имеет наряду с влиянием на гистаминовые рецепторы (возможно, Н3 – рецепторы мозга) его центральная холинолитическая активность.
4.3.2 Прометазин (дипразин, фенерган).
Обладает сильной противогистаминной активностью (более активен, чем димедрол) [13]. Дипразин является производным фенотиазина; по строению, а частично и по фармакологическим свойствам близок к аминазину. Наиболее важной фармакологической особенностью дипразина является его сильная противогистаминная (Н1-блокирующая) активность
Дипразин хорошо всасывается при приеме внутрь. При разных путях введения проникает через гематоэнцефалический барьер.
Препарат оказывает выраженное влияние на ЦНС; обладает довольно сильной седативной активностью, усиливает действие наркотических, снотворных, аналгезирующих и местноанестезирующих средств, понижает температуру тела, предупреждает и успокаивает рвоту. Он оказывает также умеренное периферическое и центральное холинолитическое действие. Сильно выражено адренолитическое действие дипразина.
4.4 М-холиноблокаторы
4.4.1 Скополамин.
Химически скополамин близок к атропину: является сложным эфиром скопина и троповой кислоты [11]. Близок к атропину по влиянию на периферические холинореактивные системы. Подобно атропину вызывает расширение зрачков, паралич аккомодации, учащение сердечных сокращений, расслабление гладких мышц, уменьшение секреции пищеварительных и потовых желез
Оказывает также центральное холинолитическое действие. Обычно вызывает седативный эффект: уменьшает двигательную активность, может оказать снотворное действие. Характерным свойством скополамина является вызываемая им амнезия.
5. Методы получения РВОТНЫХ И ПРОТИВОРВОТНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ5.1 Синтез рвотных лекарственных средств
5.1.1 Синтез апоморфина
Химический процесс [14]:
![]()
морфин апоморфин
Получение: 1 ч. чистого морфина и 10 ч. 25%-ной соляной кислоты нагревают в запаянной трубке в течение 2–3 часов при 140–150 °С. По охлаждении к содержимому трубки прибавляют избыток двууглекислого натрия и жидкость быстро взбалтывают (при возможном отсутствии воздуха) с эфиром или хлороформом. При этом неизменившийся морфин остается нерастворенным. К раствору апоморфина в эфире или хлороформе приливают небольшое количество крепкой соляной кислоты и выделившуюся хлористоводородную соль перекристаллизовывают из небольшого количества горячей воды. Из очищенной таким образом хлористоводородной соли выделяют свободное основание, прибавив к раствору соли двууглекислой соды.
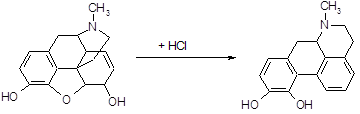
Апоморфин получается при нагревании морфина с 25 %-ной соляной кислотой в автоклаве при температуре 130–140°С в течение 2–3 часов. При этом от морфина отщепляется молекула воды [15]:
![]()
Под действием кислот разрывается кислородный мостик и этаминная цепь перемещается из положения 13 в положение 8. Происходит перегруппировка с превращением морфина в апоморфин (левовращающий). Его химическое строение отличается от строения морфина: апоморфин представляет собой почти плоскую молекулу, которую можно рассматривать как производное фенантрена и изохинолина.
Так как апоморфин-основание крайне нестоек, его применяют в виде хлористводородной соли. Поэтому полученную массу нейтрализуют содой и извлекают эфиром. К эфирной вытяжке добавляют раствор соляной кислоты и тщательно перемешивают; выделяется белый осадок хлористводородного апоморфина.
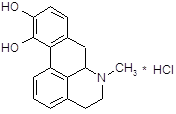
5.1.2 Синтез О, О´-диацилпроизводных апоморфина
Апоморфин в малых дозах угнетает активность дофаминергической системы и вызывает седативный эффект у животных [16]. Имеются данные об использовании апоморфина для лечения психотических нарушений при алкогольном абстинентном синдроме и шизофрении, когда наблюдается повышение активности дофаминергической системы. Однако наличие нежелательных побочных эффектов, как высокая эметическая активность и кратковременность действия, осложняют его применение в клинике. В связи с этим были проведены синтез и фармакологическое изучение некоторых О, О´-диацилпроизводных апоморфина (I – VI) с целью изыскания соединений, лишенных указанных недостатков.
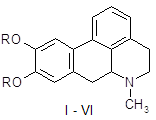
I: R = COC6H4-Br-4; II: R = COC6H4-F-4; III: R = COC6H4-CH3-4; IV: COC6H4-OPr-4; V: R = Ac; VI: R = COPh.
Экспериментальная химическая часть
ИК-спектры соединений регистрировали на спектрометре PE-580 (США) в вазелиновом масле, спектры ПМР получены на приборе «Varian» (60 МГц), внутренний стандарт – ГМДС.
О, О´-ди(4-бромбензоил)апоморфин (I). К раствору 1 г (3,2 ммоля) гидрохлорида апоморфина в смеси 3 мл диглима и 5 мл абсолютного пиридина прибавляют по каплям в токе азота 2,1 г (9,6 ммоля) хлорангидрида 4-бромбензойной кислоты. Реакционную смесь нагревают в течение 1 ч при 100°С в токе азота, выливают в 25 мл ледяной воды и эктрагируют CHCl3. Экстракт промывают насыщенным раствором NaHCO3 и NaCl, сушат безводным MgSO4 и упаривают. Получают 1,74 г I. Выходы, константы и данные спектров полученных веществ приведены в табл. 1.
Таблица 5.1
Производные апоморфина (I –IV)
| Соединение | Выход, % | Т.пл., °С | Найдено, % |
Брутто- формула |
Вычислено, % | ИК-спектр, υСО, см-1 | ||||
| С | Н | N | С | Н | N | |||||
| I | 86 | 197-8 | 58,73 | 3,78 | … | C31H232NO4 | 58,78 | 3,76 | … | 1750 |
| II | 75 | 194-6 | … | … | 2,85 | C31H23F2NO4 | … | … | 2,73 | 1750 |
| III | 81 | 184-5 | 79,02 | 5,82 | 3,13 | C33H27NO4 | 78,69 | 5,80 | 2,79 | |
| IV | 40 | 210-2,5 | 68,38 | 5,89 | 2,30 |
C37H37NO6 * * 0,5C10H8O6S2 |
68,55 | 5,61 | 1,96 | |
Примечание. Соединение V, т.пл. 124°С и VI, т.пл. 157°С. Для I найдено, %: Br 25,62. Вычислено, %: Br 25,24. Соединения I, II, IV очищены перекристаллизацией из спирта, соединение III – из CHCl3. Соотношение апоморфинового и нафталиндисульфокислотного компонентов (2:1) у соли IV подтверждено спектром ПМР.
О,О´-ди(4-фторбензоил)апоморфин (II). Из 1 г (3,2 ммоля) гидрохлорида апоморфина и 1,5 г (9,6 ммоля) хлорангидрида 4-фторбензойной кислоты аналогично соединению I получают 1,37 II.
Геми-нафталин-1,5-дисульфонат О,О´-ди(4-пропоксибензоил)апоморфина. Аналогично из 1 г (3,2 ммоля) гидрохлорида апоморфина и 1,9 г (9,6 ммоля) хлорангидрида 4-пропоксибензойной кислоты с последующей обработкой основания IV раствором нафталин-1,5-дисульфокислоты в спирте получают 0,94 г геми-нафталин-1,5-дисульфонат IV.
О,О´-ди(4-метилбензоил)апоморфин (III). Смесь 1 г (3,2 ммоля) гидрохлорида апоморфина и 5 г (32 ммоля) 4-толуиловой кислоты в 10 мл CF3COOH нагревают в течение 1 ч при 100–110°С. Реакционную смесь упаривают в вакууме, остаток встряхивают с 50 мл эфира и 30 мл насыщенного раствора NaHCO3. Эфирный раствор отделяют, промывают водой, упаривают и получают 1,04 г III.
5.1.3 Синтез бромокриптина
Промышленные методы получения алкалоидов спорыньи, применяемые за рубежом, основаны на извлечении их из предварительно обезжиренной спорыньи органическими растворителями [17].
В Харьковском научно-исследовательском химико-фармацевтическом институте предложен метод избирательной водной зкстракции алкалоидов из спорыньи, в результате которой отдельно получают зкстракты, содержащие эргометрин, и экстракты, содержащие полипептидные алкалоиды. Первые используют для получения из них зргометрина, вторые – для выделения эрготоксина и эрготамина.
Экстракты, содержащие алкалоиды полипептидного типа, прозрачны, слабо окрашены; алкалоидов в них содержится 0,2–0,4 мг/мл (в зависимости от содержания их в исходной спорынье), экстрактивных веществ – 0,2–0,3 %, рН экстрактов – около 2,0.
Таблица 5.2
Алкалоидный состав ряда образцов ржаной спорыньи
| Место сбора спорыньи | Общее содержание алкалоидов (в % к весу спорыньи) | Группа | Правовращающие группы эрготоксина | |||
| эрготамина | эрготоксина | |||||
| эрготамин | эрготаминин |
эргокристин + эргокорнин |
эргокриптин | |||
| в % к общей сумме алкалоидов | ||||||
| Киевская область | 0,140 | 17,5 | Следы | 36,3 | 17,5 | 11,7 |
| » » | 0,266 | 20,0 | 0 | 60,6 | 0 | 0 |
| Харьковская область | 0,300 | 16,8 | Следы | 49,1 | Следы | 3,6 |
Примечание. Все данные приведены в пересчете на эргокристин. Общее содержание алкалоидов определяли по водному методу, отдельные алкалоиды – методом хроматографии на бумаге.
Алкалоидный состав зкстрактов устанавливали методом хроматографии на бумаге в системе бензол формамид. Он соответствовал составу исходной спорыньи. Исходной спорыньей служили разные образцы дикорастущей ржаной спорьньи. В таблице приведен алкалоидный состав некоторых наиболее характерных образцов.
Алкалоиды выделяли из экстрактов по схеме получения эрготала. Алкалоиды высаливали добавлением 20–25 % раствора хлористого натрия. Из выпавшего осадка их извлекали хлороформом в щелочной среде и после концентрирования хлороформных экстрактов осаждали петролейным эфором. Выделенный при этом продукт представлял собой смесь нерастворимых в воде алкалоидов с [a]D20 = –20°, –60° (с 1, хлороформ). Эту смесь обрабатывали фосфорной кислотой в ацетоновом растворе с целью перевода ее полностью в левовращающие, физиологически активные алкалоиды. Полученные фосфорнокислые соли алкалоидов переводили затем в основания путем обработки их бикарбонатом натрия в водной среде с последующей экстракцией хлороформом. Из сгущенных хлороформных экстрактов осаждали алкалоиды в форме тартратов прибавлением 5 % раствора винной кислоты. Выпавшие тартраты смешивали с окисью магния. Выделенные таким образом основания экстрагировали хлороформом и после сгущения хлороформных экстрактов осаждали петролейным эфиром. Полученную смесь оснований растворяли в бензоле и хроматографировали на колонке с окисью алюминия. Бензолом элюировали алкалоиды эрготоксиновой группы, затем хлороформом эрготамин.
Выделение алкалоидов группы эрготоксина. При упаривании бензольных элюатов выпадал кристаллический эрготоксин в виде комплексного соединения с бензолом с [a]D20 = –130°, –160° (с 1, хлороформ). Анализ его на бумажной хроматограмме в системе бензол – формамид показал, что он содержит в основном эргокристин, эргокриптина было меньше или он вовсе отсутствовал. Поскольку эргокорнин в этой системе располагался на хроматограмме на уровне эргокристина, дополнительно производился анализ косвенным путем: хроматографии на бумаге в системе бутанол – уксусная кислота вода (4:1:5) подвергались аминокислоты, полученные в результате кислотного гидролиза эрготоксина. Присутствие валина в кислотном гидролизате должно было бы указать на содержание в исследуемом соединении эргокорнина. Результаты анализа показали, однако, что в большинстве полученных образцов комплекса эрготоксин – бензол эргокорнин отсутствовал; это хорошо согласуется с данными, полученными нами ранее, при исследовании алкалоидного состава дикорастущей спорыньи отечественного происхождения.
При перекристаллизации комплекса эрготоксин – бензол из ацетона выпадало крнсталлическое основание чистого эргокристина в соединении с ацетоном, [a]D20 = –165°, –187° (с 1, хлороформ). Это основание было обозначено в дальнейшем как эргокристин-ацетон. Выход его составлял около 60 % взятого для кристаллизации комплекса эрготоксин бензол.
В ацетоновых маточниках оставался частично эргокристин, а также эргокриптин, если он первоначально обнаруживался в комплексе эрготоксин – бензол. Все наши попытки разделить эргокристин н эргокриптин фракционной кристаллизацией из разных растворителей не привело к положительным результатам.
Для разделения этих алкалоидов воспользовались методом фракционной кристаллизации их в виде солей ди-(n-толуил)-l-винной кислоты. Разделению подвергали первоначальный комплекс эрготоксин – бензол и ацетоновые маточники, полученные после отделения эргокристин-ацетона.
При фракционной кристаллизации в первую очередь выпадала кристаллическая нейтральная соль эргокристина, а затем эргокриптин в форме кислой соли.
Бромирование эргокриптина. Получают 2-бром-α-эргокриптин бромированием α-эргокриптина – основного алкалоида спорыньи эргокриптинового штамма [18]. Описано несколько методов бромирования с использованием различных бромирующих агентов: брома, N-бромсукцинимида (NБС), N-бромкапролактама, диоксандибромида, N-бромфталимида, гидротрибромида пирролидона-2, бромсахарина, гидротрибромида пиперидона-2 и др. Однако эти методы, как правило, либо сложны для промышленного исполнения, либо дают невысокий выход 2-бром-α-эргокриптина. Так, наиболее распространенный метод бромирования NБС дает выход продукта менее 50 %
Целью настоящей работы является поиск более эффективных методов бромирования В качестве исходного материала мы использовали индовидуальные α- и β-эргокриптины, а также более доступную смесь α- и β-эргокриптинов без предварительного их разделения, Эту задачу решали либо путем модернизации известного метода бромирования NБС, либо путем применения новых для этой реакции бромирующих агентов 2,4,4,6-тетрабромциклогексадиен-2,5-она (ТБЦГ), 2,2-дибром-5,5-диметилцикло-гександиона-1,3 (дибромдимедон), пербромида фенилтриметил-аммония (ФТМА) и «полимерного пербромида» на основе амберлита ИРА-402.
Экспериментальная часть
Контроль реакционных смесей осуществлялся хроматографически на пластинках «Silufol UV-254» в системе СН2Сl2 – диоксан – этиловый спирт – аммиак конц., 36:3:1:0,2, или на пластинках «DC-Alufolien» с нейтральной Al2O3 60F254 Typ E (ФРГ) в системе бензол – CHCl3, 1:1. Определение выхода 2-бромэргокриптинов при кинетических исследованиях проводили путем контроля на хроматографических пластинок на денситометре «Сromoscan 200» (Jouce Loebl).
Для изучения реакций бромирования в качестве исходного субстрата использовали техническую смесь изомеров α- и β-эргокриптинов состава 60:40 % соответственно, а также индивидуальные изомеры.
Полимерный пербромид на основе ионобменной смолы амберлит ИРА-402. Смешивают 10 г ионобменной смолы амберлит ИРА-402 с 5 % раствором KBr так, чтобы смола была полностью покрыта слоем раствора, и оставляют для набухания на ночь. Затем сливают верхний водный слой, смолу заливают свежим раствором KBr и приливают по каплям 2 мл брома при непрерывном перемешивании. Полученный бромирующий агент отфильтровывают, промывают водой, затем сухим диоксаном. Сушат сначала над CaCl2 в вакуум-эксикаторе, а затем над P2O5.
Бромирование эргокриптинов NБС. К нагретому до 60°С раствору 1 г (1,74 ммоля) смеси изомеров α- и β-эргокриптинов в 20 мл абс. диоксана (перегнанного над бензофенонкетилнатрием) в атмосфере азота и в темноте (или в зачерненной снаружи колбе) добавляют по каплям в течение 5 мин при перемешивании раствор 0,37 г (2,04 ммоля) NБС в 6,5 мл абс. диоксана. Реакционную смесь перемешивают в этих же условиях еще 70 мин, охлаждают, диоксан упаривают в вакууме при 40–50°С. Остаток растворяют в 30 мл СН2Сl2, полученный раствор промывают 20 мл 2 н. раствора Na2CO3. Водную фазу экстрагируют СН2Сl2 (2×10 мл). Объединенные органические фазы промывают 50 мл воды, сушат над Na2SO4, растворитель упаривают. Получают 1,1 г темного вязкого масла, которое очищают хроматографически на колонке (10×2 см) с Al2O3 III ст. активности, элюируя последовательно бензолом, смесью бензол – CHCl3, 4:1, 3:2, 1:1. Получают 625 мг (55 %) смеси α- и β-2-бромэргокриптинов. Полученный образец хроматографически идентичен с образцами, полученными при хроматографировании на силуфоле, Al2O3 («Merek»), а также методом ВЭЖХ.
Бромирование эргокриптинов 2,4,4,6-тетрабромциклогексадиен-2,5-оном. К нагретому до 60°С раствору 575 мг (1,0 ммоль) смеси α- и β-эргокриптинов в 20 мл абс. диоксана при перемешивании добавляют сразу раствор 410 мг (1,0 ммоль) тетрабромциклогексадиенона в 10 мл абс. диоксана. Реакционную смесь перемешивают в течение 30 мин при 60°С. Охлаждают, диоксан упаривают в вакууме, остаток растворяют в 30 мл СН2Сl2. Полученный раствор промывают 20 мл 5 % раствора NaHCO3, затем водой и сушат над Na2SO4. Растворитель упаривают в вакууме, остаток очищают хроматографически на колонке (10×2 см) с Al2O3 III ст. активности, элюируя последовательно смесью бензол – CHCl3, 3:1, 3:2, 1:1. Получают 382 мг (85 %) смеси α- и β-2-бромэргокриптинов.
Бромирование эргокриптинов пербромидом фенилтриметил-аммония. К раствору 575 мг (1,0 ммоля) смеси α- и β-эргокриптинов в 30 мл абс. СН2Сl2 (перегнанного над P2O5) при перемешивании добавляют 2 г мелко измельченной и предварительно высушенной при 120°С MgO. К полученной суспензии при перемешивании добавляют в течение 30 мин раствор 320 мг (0,85 ммоля) пербромида фенилтриметиламмония в 30 мл абс. СН2Сl2. В процессе добавления пербромида к реакционной смеси через каждые 5–7 мин вносят свежие порции MgO по 50–100 мг. Реакционную смесь фильтруют, осадок MgO тщательно промывают несколько раз 40 мл СН2Сl2, фильтрат промывают 20 мл 5 % раствора NaHCO3. Водный слой дополнительно экстрагируют СН2Сl2 (2×10 мл). Объединенные органические фракции промывают водой и сушат над Na2SO4. Растворитель упаривают в вакууме. Остаток растворяют в минимальном количестве бензола и наносят на хроматографическую колонку (16×2 см), заполненную Al2O3 IV ст. активности. Элюируют последовательно бензолом, смесью бензол – CHCl3, 5:1, 4:1, 3:1, 1:1. Получают 460 мг (70 %) смеси α- и β-2-бромэргокриптинов. Полученный образец идентичен образцам, полученным при хроматографировании на силуфоле, Al2O3 и методом ВЭЖХ.
Бромирование эргокриптинов «полимерным пербромидом» на основе амберлита ИРА-402. Растворяют 2 г α- и β-эргокриптинов в 40 мл абс. СН2Сl2, добавляют 4 г сухой MgO и при перемешивании в течение 0,5 ч вносят 6 г вышеуказанной смолы. Премешивают в течение еще 75 мин. После чего растовр отделяют от MgO и смолы. Раствор обрабатывают 40 мл 2 н. Na2CO3, затем водой (2×20 мл), органические фазы объдиняют, сушат Na2SO4, фильтруют и упаривают. Получают 2,1 г технического продукта, который очищают на колонке с Al2O3 III ст. активности в соотношении 1:30. Алкалоиды элюируют бензолом, смесью бензол – CHCl3 (5–30 %). Получают 0,4 г (20 %) смеси α- и β-2-бромэргокриптинов.
5.2 Синтез противорвотных лекарственных средств
5.2.1 Синтез аминазина
2-Хлорфенотиазин основной полупродукт синтеза ценного и широко применяемого лечебного препарата аминазина [19]. Кроме того, он является основным полупродуктом синтеза другого лечебного препарата – хлорацизина, применяемого при лечении заболеваний сердечно-сосудистой системы.
Наиболее рациональноной схемой получения 2-хлорфенотиазина является конденсация 3-хлордифениламина с серой (сплавление) в присутствии йода.

Реакция протекает с образованием двух изомерных хлорфенотиазинов – 2-хлорфенотиазина и 4-хлорфенотиазина.
Впервые эта реакция описана Шарпантье и Жолио. Авторы проводили конденсацию при небольшом избытке серы в присутствии 1 % йода при 180°С. Они приводят данные о температуре плавления образующихся продуктов без описания метода разделения и указания выхода 2-хлорфенотиазина.
Н. В. Савицкая и соавторы разработали условия конденсации 3-хлордифениламина с серой и очистки плава хлорфенотиазина. Реакцию 3-хлордифениламина с небольшим (2 % теоретического количества) избытком серы проводят в присутствии йода (1 % веса 3-хлордифениламина). Реакционную массу нагревают до 175–180°С и выдерживают при этой температуре до прекращения выделения сероводорода. Плав после кипячения в хлорбензоле с активированным углем фильтруют. Выкристаллизовавшийся и отфильтрованный хлорфенотиазин промывают хлорбензолом и спиртом. Выход 2-хлорфенотиазина составляет 56,2 %, считая на 3-хлордифениламин. Температура плавления 2-хлорфенотиазина 191–194°С.
В производстве аминазина получение 2-хлорфенотиазина является наиболее трудным процессом, что обуловлено относительно высокой температурой сплавления (175–180°С), образованием изомера – 4-хлорфенотиазина, смолистых веществ и выделением больших количеств сероводорода. В производственных условиях в силу ряда причин осмоление более значительно, чем в лабораторных условиях, поэтому даже сравнительно невысокий выход 2-хлорфенотиазина (56 %) в производстве практически не достигается.
Все эти обстоятельства заставили нас искать путей смещения равновесия реакции с целью повышения выхода 2-хлорфенотиазина. Попытки вести реакцию в каких-либо наиболее доступных и обычно применяемых органических растворителях не привели к положительным результатам. Лучшей средой для ведения реакции оказалось само основное вещество – 3-хлордифениламин. Мы установили, что проведение реакции 3-хлордифениламина с серой при избытке 3-хлордифениламина и в присутствии остатка после отделения 2-хлорфенотиазина, содержащего главным образом 4-хлорфенотоазин, смещает реакцию в сторону преомущественного образования 2-хлорфенотиазина.
Экспериментальная часть
В трехгорловую круглодонную колбу, снабженную мешалкой и термометром, помещают 100 г перегнанного 3-хлордифениламина (0,49 г/м), 20 г серы (0,31 г/м) и 1 г йода (0,005 г/м). Полученную смесь нагревают при перемешивании до 170–180°С и выдерживают при этой температуре до прекращения выделения сероводорода (1 – 1½ часа). По окончании выдержки к реакциовной массе прибавляют 175 мл хлорбензола и 5 г угля, кипятят в течение 15 минут (130–132°С) и затем фильтруют горячий раствор, освобождая его от угля и смолистых примесей. Фильтрат охлаждают до 5–10°С и оставляют на 3–4 часа для кристаллизации. Вьипавший осадок 2-хлорфенотиазина отфильтровывают, промывают небольшим количеством хлорбензола и этилового спирта и сушат при 100°С. Получают 42–45 г 2-хлорфенотиазина с температурой плавления 195–200°С (в пределах 1°С).
От маточного раствора после отделения 2-хлорфенотиазина отгоняют спирт и хлорбензол, к остатку прибавляют 32 г 3-хлордифениламина, 10 г серы и 0,5 г йода и повторяют синтез, как описано выше. Подобным же образом поступают с остатками после отделения 2-хлорфенотиазина в последующих 9–10 опытах с той только разницей, что нагревают до 175–180°, а не до 150–160°, как в первых двух опытах.
В итоге при употреблении в цикле из 11 опытов 420 г 3-хлордифенил-амина получают 380 г 2-хлорфенотиазина, что составляет 79 % теоретического выхода, считая на израсходованный 3-хлордофениламин.
Количество 3-хлордифениламина (32 г) во втором и последующих опытах определяется весом получаемых остатков после отделения 2-хлорфенотиазина с учетом того, чтобы сумма веса 3-хлордифениламина и остатка была близка к первоначальной загрузке, т. е. к 100 г.
Поскольку в цикле находится избыток 3-хлордифениламина, количество взятой серы близко к теоретическому (по отношению к вновь загружаемому 3-хлордифениламину).
Вес остатка не увеличивается с увеличением числа проведенных опытов, что подтверждает предположение о смещении равновесия реакции в сторону образования 2-хлорфенотиазина, когда в остатке уже имеется изомер 4-хлорфенотиазин, так как вновь загружаемый 3-хлордифениламин расходуется главным образом на образование 2-хлорфенотиазина, иначе с увеличением числа проведенных опытов должен был бы существенно увеличиваться вес остатка.
Рециркуляция остатка длится до тех пор, пока не истощится избыток 3-хлордифениламина. При 10-й рециркуляции остатка 2-хлорфенотиазин имеет точку плавления 193–194°С, окраска хлорфенотиазина темно-зеленаяж. Дальнейшая рециркуляция уже нецелесообразна, так как наступает еще более резкое понижение качества.
Мы провели серию опытов, в которой, начиная с 3-го опыта, брали по 35 г 3-хлордифениламина т. е. на 10 % больше, чем в предыдущих опытах. В этом случае удалось вести рециркуляцию остатка до 20-го опыта, но выход остался на прежнем уровне (77 %). Поэтому соотношение компонентов реакции в первой серии опытов следует считать наиболее рациональным.
Описанный способ позволяет получать 2-хлорфенотиазии высокого качества, с температурой плавления 195–200°С (в пределах 1°С) и выходом 79 % теоретического, считая на 3-хлордифениламин.
Заключительной стадией в синтезе аминазина (I*HCl) является алкилирование 2-хлорфенотиазина (II) диметиламинохлорпропаном (III) [20]. Подобран оптимальный режим процесса: реагенты II и III кипятят в смеси толуола с хлорбензолом в присутствии порошкообразного едкого натра, причем выход I достигает 90 %. Проведение реакции аналогичным образом, но в присутствии катализатора межфазного переноса (КМФ) не дает очевидных преимуществ; выход I составляет 78 %. В более мягких условиях, типичных для реакции межфазного переноса (80°С, 25 мол.% КМФ, бензол/водный раствор NaOH) соединение I получить не удалось, хотя в тех же условиях фенотиазин II алкилируется хлористым бензилом и бромистым этилом. Низкая эффективность III как алкилирующего агента была отмечена, но не получила объяснения.
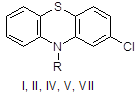
I: R = (CH2)3NMe; II: R = H; IV: R = CH2Ph; V: R = Me; VII: R = CH2CH=CH2
Экспериментальная часть
ГЖХ-анализы проведены на хроматографах «Varian-3700» (США) и «Chrom-5» (ЧССР) с пламенно-ионизационным детектором; колонки стеклянные или стальные (100×0,3 см), заполненные сорбентом (5% OV-17) на хроматоне «N-super». Замена стеклянных колонок на металлические, а также изменение температуры испарителя (210–350°С) не приводит к термокаталитическому разложению основания аминазина I: на хроматограммах не меняется соотношение пиков и не появляются дополнительные пики. Режим анализа: температура анализа колонок 210°С, испарителя 250°С, детектора 250°С, скорость газа-носителя (азот) 30 мл/мин, время удерживания веществ V, VII и I 3, 3,8 и 7 соответственно.
Масс-спектры электронного удара получены на хромато-масс-спектрометре МАТ-112 («Varian», ФРГ), энергия ионизирующих электронов 70 эВ. Ввод образца реакционной массы осуществлялся через хроматограф «Varian Aerograph 1440»; колонки и режим их работы такие же, как и при ГЖХ-анализе.
Гидрохлорид 2-хлор-10-(γ-диметиламинопропил)-фенотиазина (I*HCl) и 2-хлор-10-метилфенотиазин (V). Реакцию II (0,05 моль) и III проводят согласно [1], продолжительность отгонки воды 4,5 ч. Далее реакционную смесь промывают водой, смесь растворителей отгоняют в вакууме, остаток сушат до постоянной массы (16,13 г) и анализируют методом ГЖХ.
Полученную смесь растворяют в 70 мл толуола, прибавляют раствор HCl в этаноле, раствор упаривают в вакууме до объема 30 мл, выпавшие кристаллы отфильтровывают и после перекристаллизации из смеси толуол–изопропиловый спирт (4:1) получают 10,92 г I*HCl (61,4%).
Маточный раствор после отделения технического I*HCl промывают водой, толуол отгоняют, остаток тщательно сушат в вакууме и анализируют на хромато-масс-спектрометре. Масс-спектр m/z (IОТН): V – 247(100), 232(65), 215(12), 212(19); VII – 273(11), 247(8), 233(95), 232(100), 198(39). После двух перекристаллизаций из гексана получают 0,1 г V, т.пл. 81–83°С.
5.2.2.1 Синтез анестезина из р-нитротолуола
В качестве исходного сырья для получения этого продукта лучше всего применять р-нитротолуол, во-первых, потому, что его легко и дешево можно купить, и, во-вторых, потому, что отдельные фазы получения через этиловый эфир р-нитробензойной кислоты до эфира р-аминобензойной кислоты дают хорошие выхода и протекают гладко, хотя и требуют частично довольно продолжительного времени для завершения реакции [21]:
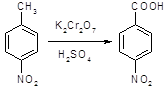
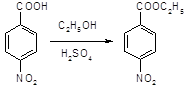
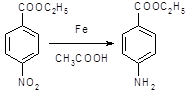
р-Нитробензойная кислота из р-нитротолуола. В гомогенно освинцованном двустенном котле с крышкой, мешалкой, обратным холодильником и трубкой для выдавливания большого внутреннего диаметра заливают 200 л воды, к которым при помешивании и охлаждении водой медленно приливают 150 кг технической серной кислоты 66° Вё. По охлаждении прибавляют 60 кг двухромовокислого калия и 15 кг р-нитротолуола, закрывают люк и нагревают до кипения 24 часа при помешивании с обратным холодильником. Перемешивают для того, чтобы нитробензойная кислота не обволакивала нитротолуола и не мешала окислению. Кипение способствует окислению; при температуре ниже кипения окисление протекает медленнее. Охлаждающая вода в обратном холодильнике должна подогреваться острым или глухим паром до 40–45°С, для того чтобы увлекаемый водяными парами р-нитротолуол не застыл там и не вызвал закупоривания. По окончании окисления выдавливают реакционную массу при 60–70°С на нучу. Последняя изготовляется из гомогенно освинцованного железа; для фильтрования служит кислотоупорная фильтровальная плитка. Горячие растворы тотчас спускают в эмалированные или же в гончарные сосуды. Из них в течение нескольких дней кристаллизуется большая часть взятого хрома в виде хромовокалиевых квасцов. Нитробензойную кислоту промывают на нуче до исчезновения серной кислоты, растворяют в разбавленном растворе соды, раствор фильтруют через мешок и осаждают технической соляной кислотой, отсасывают на нуче, промывают водой и высушивают при 40–50°С.
Выход достигает самое меньшее 16 кг.
Этиловый эфир р-нитробензойной кислоты. Для получения его берут тот же аппарат, как и для р-нитробензойной кислоты, только кроме обратного холодильника нужен еще и прямой холодильник.
В аппарат загружают 16 кг сухой р-нитробензойной кислоты, заливают ее четырехкратным по весу количеством 96 %-ного этилового спирта, растворяют при помешивании, прибавляют 8 кг технической серной кислоты 66° Вё и нагревают без помешивания 16–20 часов с обратным холодильником до кипения.
После охлаждения нейтрализуют при открытом люке медленно и осторожно водным раствором соды; для нейтрализации необходимо 11–12 кг соды Сольвэ. Отгоняют спирт с прямым холодильником, по охлаждении выдавливают остаток на глиняную нучу и промывают на последней эфир водой.
Из фильтрата выделяют не вошедшую в реакцию нитробензойную кислоту технической соляной кислотой, отсасывают на нуче, промывают водой и высушивают.
Выход 13,5–14,5 кг чистого эфира. Остаток получают в виде неизмененной кислоты обратно.
Этиловый эфир р-аминобензойной кислоты. Чтобы получить и сырой продукт по возможности в чистом виде, раньше восстанавливали нитроэфир порошком олова и соляной кислотой. Полученную смесь после восстановления разбавляли большим количеством воды, точно нейтрализовали едким натром, нагревали до кипения и горячий водный раствор основания анестезина отделяли фильтрованием через небольшой фильтрпресс от выпавшего гидрата окиси олова. Технический анестезин, выпавший по охлаждении фильтрованного раствора, был достаточно чист. Маточники его можно было применять вместо чистой воды при новых операциях. Полученный таким образом технический продукт можно было превратить в чистое основание только одной перекристаллиацией из 80 %-ного спирта. Пасту гидрата окиси олова с фильтрпресса можно было продать, как таковую, или восстановить углем в металлическое олово в вагранках.
Этот метод восстановления был бы хорош при условии, если бы он проводился в свободной от железа аппаратуре и технический продукт можно было очень легко очищать. Анестезин при соприкосновении с железом краснеет, так же как и калий сульфогваяколовый. Но финансовые убытки при продаже пасты окиси олова были значительны, а регенерация олова в вагранках для сравнительно небольших количеств была сложна.
Поэтому в настоящее время повсюду восстановительный процесс ведут железом и уксусной кислотой и полученный таким образом технический продукт перед кристаллизацией перегоняют в вакууме. Анестезин можно перегонять уже при обыкновенном давлении при 305°С, но при этом к концу операции невозможно избежать разложения. Напротив, при остаточном давлении в 10–20 мм получается почти белый, слегка желтоватый продукт.
Для восстановления железом и уксусной кислотой применяют аппарат, для которого достаточно объема в 200 л. Загружают в него 20 кг обезжиренных и просеянных чугунных опилок и 10 л воды. и травят при 70°, при помешивании 4 кг 80%-ной уксусной кислоты. Затем останавливают приток пара и при непрерывном помешивании постепенно приливают в аппарат 16 кг этилового эфира р-нитробензойной кислоты. Реакция сильно экзотермична, и вода выпаривается, хотя аппарат не подогревают. Время от времени приливают свежей воды взамен испаряющейся. По прилитии всего эфира, что берет 1½ – 2 часа, продолжают помешивание при легком подогревании еще 2 часа. Восстановление закончено, когда капли жидкости от восстановления более не окрашивают пропитанной раствором соды фильтровальной бумаги. Тогда дают жидкости остыть и нейтрализуют на лакмус раствором соды. Выделившееся при этом в свободном состоянии основание анестезина извлекают при 60° три раза, каждый раз 50 кг бензола. Последнюю вытяжку сохраняют, чтобы употребить вместо свежего бензола для первого извлечения следующей порции.
Из двух первых бензольных вытяжек, высушенных хлористым кальцием, отгоняют бензол и перегоняют остаток сильно окрашенное основание технического анестезина, – в вакууме. Служащий для этого маленький аппарат лучше всего делать из чистого алюминия; для наблюдения за перегонкой служит надставка из стекла или лучше из плавленого горного хрусталя. Прерывают перегонку, когда погон не стекает больше бесцветным. Нагревание можно производить маленькой газовой печью.
Погон перекристаллизовывают из 80%-ного спирта. Для превращении основания, которое для этого должно быть чисто белым, в его хлоргидрат, растворяют его на холоду в абсолютном спирте и осторожно и медленно нейтрализуют раствор на конго 30%-ной спиртовой соляной кислотой. На следующий день выделившийся солянокислый анестезин отсасывают на нуче, промывают небольшим количеством холодного абсолютного спирта и высушивают при З0 – 40°.
Выход по способу восстановления железом и уксусной кислотой бывает не менее 80% теории.
5.2.2.2 Синтез анестезина из р-толуидина
Другим исходным продуктом для анестезина является р-толуидин. Он переводится в ацет-р-толуидин, а последний окислением марганцовокислым калием – в р-ацетаминобензойную кислоту. Эта кислота с одновременным образованием уксусного эфира переходит в этиловый эфир р-аминобензойной кислоты:
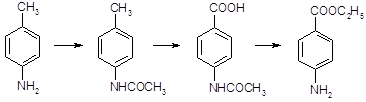
Ацетилирование. В колбе Шотта емкостью 15 л нагревают на масляной бане 5 кг р-толуидина и 6,5 кг 80%-ной уксусной кислоты в течение 12 часов до 140°. На колбу устанавливают широкую восходящую трубку, к которой присоединяют прямой холодильник. Нагревают очень медленно. Избыток уксусной кислоты отгоняется. Если последние часы держать температуру 135 – 140°, то ацетилирование оканчивается в 12 час. Содержимое колбы выливают в 25 л воды, выделившийся технический ацетат отсасывают на нуче и высушивают.
Выход: 5,5 кг ацет-р-толуидина.
Окисление. В деревянном чану емкостью 600 л с мешалкой и паровым змеевиком нагревают 500 л воды и 11 кг мелко порошкованного ацет-р-аминотолуидина до 30° и к раствору прибавляют сначала 8 кг мелко истолченного марганцевокислого калия. Температура сама поднимается до 40°. Небольшим подогреванием паром ее поднимают до 45°. Через 2 часа прибавляют еще 8 кг марганцевокислого калия и через 3 следующие часа – третью порцию, тоже 8 кг, так что всего уходит 24 кг марганцевокислого калия. После того как весь марганцевокислый калий прибавлен, перемешивают еще некоторое время, затем останавливают мешалку и дают осесть осадку перекиси марганца.
Прозрачный раствор сливают, осадок переносят в мешки и отмывают его там от ацет-р-аминобензойной кислоты. Наконец осадок еще фугуют. Все растворы соединяют, и еще раз фильтруют. Из фильтрата осаждают прибавлением разбавленной серной кислоты до кислой реакции на конго, фильтруют, промывают и высушивают.
Выход: 13 кг ацет-р-аминобензойной кислоты.
Отщепление ацетильной группы и одновременное образование эфира. В колбе Шотта емкостью 15 л кипятят 1 час с обратным холодильником 3 кг ацет-р-аминобензойной кислоты с 6,8 кг 96%-ного спирта и 2,4 кг серной кислоты 66° Вё. После этого все выливают в 40 л воды и постепенно доводят поташом до щелочной реакции на фенолфталеин. Технический эфир отсасывают на нуче и высушивают. Он имеет желтоватый оттенок, который, если продукт не хотят перегонять в вакууме, можно удалить только тем, что его растворяют в десятикратном количестве воды с помощью свободной от железа соляной кислоты с прибавкой небольшого количества сернистой кислоты и угля для обесцвечивания, свободного от металла, и раствор фильтуют. Фильтрат снова осаждают поташом. Этот процесс растворения и осаждения иногда приходится повторять несколько раз. Наконец, предварительно очищенный таким образом эфир перекристаллизовывают из разбавленного спирта. Растворяют 5 кг технического эфира в 7 л 50%-ного спирта. Выкристаллизовывается 85 – 90 % эфира. Маточники можно использовать 1 – 2 раза для перекристаллизации, после чего из них отгоняют спирт. Из водного остатка выделяется технический эфир, который после промывания высушивают и снова перекристаллизовывают.
Выход: 2,5 кг анестезина.
5.2.2.3 Изучен метод получения анестезина гидрированием этилового эфира n-нитробензойной кислоты на палладиевых катализаторах [22].
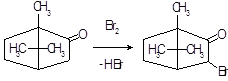
Производство монобромкамфоры рентабельно потому, что в виде побочного продукта получается бромистоводородная кислота, имеющая большой спрос и потому довольно дорогая [21].
Аппаратура для бромирования аналогична применяемой при производстве трибромфенола.
Рис. 5.3 Аппаратура для бромирования трибромфенола.
Так как загрузки делают едва ли больше 10 кг, то достаточна глиняная турилла емкостью 50 л и соответствующий глиняный горшок для улавливания бромистого водорода. Глиняную туриллу хорошо заменить фарфоровой и нагревать нужно не горячей водой, а на масляной бане, обогреваемой паром.
Исходным продуктом является чистая естественная камфора, а не синтетическая. Последняя, даже если она относительно чиста, всегда дает худшие выхода, чем естественная камфора.
В фарфоровую туриллу вносят 10 кг порошкованной камфоры и при частом помешивании толстой стеклянной палкой приливают в течение 3 часов точно 10,45 кг брома. Вследствие медленного приливания брома его можно производить из маленькой делительной воронки.
Затем смесь нагревают в течение 3 часов до 80°С, поднимая после этого температуру в продолжение 3 часов до 100°С. Выделяющийся бромистый водород улавливается дистиллированной водой.
По окончании выделения бромистого водорода выдавливают плав из фарфоровой туриллы в глиняный горшок, в который налит предварительно профильтрованный 5%-ный раствор соды. Помешивают до тех пор, пока плавающая на поверхности техническая бромкамфора не застынет в крупитчатую массу. Ее отсасывают на нуче и промывают холодной дистиллированной водой, пока промывные воды не перестанут реагировать с азотнокислым серебром. Водные растворы идут на приготовление бромистого натрия; техническую бромкамфору сушат при 35°С.
Растворяют 3 части бромкамфоры в 4 части горячего 95 %-ного спирта, прибавляют к горячему раствору немного свободного от металлов обесцвечивающего угля и через 10 минут фильтруют через складчатый фильтр в большую колбу Эрленмейера для кристаллизации. Уже при обыкновенной температуре кристаллизуется около 50 % бромкамфоры в виде бесцветных иголок. Для усиления кристаллизации колбу ставят на несколько часов в смесь льда с солью, отсасывают кристаллы на нуче, споласкивают небольшим количеством спирта и сушат при 35°С.
Маточник с промывным спиртом упаривают на водяной бане до ¼ объема и опять дают кристаллизоваться. Эту операцию повторяют еще раз. Последние довольно темные растворы от различных операций собирают и перерабатывают вместе. Их разбавляют полуторным по весу количеством дистиллированной воды, вследствие чего выделяется мазеобразное вещество. Его отделяют от прозрачного раствора, от которого отгоняют спирт. Из охлажденного остатка от дистилляции выделяется техническая бромкамфора, которую сушат и присоединяют к прочей технической бромкамфоре для очистки перекристаллизацией.
Мазеобразное вещество сушат и определяют его точку плавления. Она обычно выше вследствие примеси непробромированной камфоры. Тогда продукт бромируют, беря 4–5 % от его веса брома, и опять определяют температуру плавления. Если она удовлетворительна, то продукт присоединяют к технической бромкамфоре. Если точка плавления этого вещества низка, то значит оно содержит дибромкамфору. Тогда его кипятят ½ часа с обратным холодильником с 10 % от его веса щелочи, выливают плав в воду, фильтруют, отмывают от щелочи и после сушки опять определяют температуру плавления.
Выход: 122 % от взятой в работу камфоры, если это была чистая естественная камфора. Из синтетической камфоры выход тем ниже, чем меньше ее чистота.
Ментиловый эфир валериановой кислоты можно приготовить пропусканием сухого хлористого водорода через смесь из моногидрата валериановой кислоты и ментола [21].
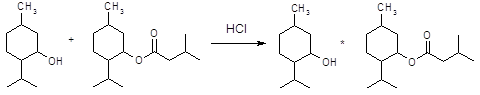
В достаточной величины круглодонную колбу из иенского стекла или пирекс, стоящую на паровой бане, вносят: 5,1 кг моногидрата валериановой кислоты и 7,8 кг ментола и пропускают в смесь сухой хлористый водород сначала 4 часа без нагрева, затем начинают постепенно нагревать, все время продолжая пропускать хлористый водород. Берут изредка пробы, взбалтывают с водой, нейтрализуют содой и смотрят, насколько прошел процесс этерификации. Когда он окончен (примерно через 7 часов пропускания хлористого водорода), смесь промывают в одной или нескольких склянках с тубусом у дна. Сначала основательно промывают водой, пока промывная вода почти не перестанет давать реакции с азотнокислым серебром. Затем следует промывка очень разбавленным раствором едкого натра (но не содой, так как вследствие выделения углекислоты в склянках может создаться избыточное давление, что опасно и может привести к потерям!). Сначала взбалтывают ½ мин., самое большее 1 мин., с маленьким избытком раствора щелочи, затем быстро отделяют раствор и опять промывают водой. Разбавленная щелочь и даже вода действуют на эфир омыляюще, хотя и медленно. Взбалтывание и отделение обоих слоев должно поэтому производиться очень тщательно.
Отмывка от кислоты и последующая сушка обезвоженной глауберовой солью должны производиться с самим эфиром, а не с его раствором в эфире, бензоле или каких-либо других органических растворителях, так как они могут изменить запах продукта в неблагоприятную сторону.
Полученный эфир должен быть обязательно перегнан в высоком вакууме при 3–5 мм остаточного давления. Эта операция необходима, иначе нельзя быть уверенным, что он на свету не пожелтеет. Он кипит при указанном вакууме пи 165–170°С. При перегонке его нельзя нагревать голым огнем горелки или на песчаной бане, надо на масляной бане, во избежание местных перегревов, которые вредно отражаются на запахе.
После перегонки в вакууме определяют число омыления Кетсторфа. Для чистого продукта это число равняется 233. Нужно иметь в виду, что полное омыление продолжается очень долго – около 4 часов.
Продажный Mentholum valerianicum имеет число омыления 162, так как содержит до 30 % свободного ментола. Посредством растворения в нем ментола продукт устанавливают на это число омыления, для чего ментола обычно идет 25–30 % от веса эфира.
Выход из указанной загрузки: чистого эфира 11–11,5 кг, и продажного Mentholum valerianicum 14–14,5 кг.
Чтобы продукт на свету оставался бесцветным не обладал чисты запахом, все исходные материалы должны быть возможно чище.
Ментол должен быть фармакопейного качества. Чистая валериановая кислота очень дорога, поэтому берут техническую валериановую кислоту и очищают сами. Для очистки ее перегоняют из большой кварцевой или стеклянной реторты через колонку, наполненную стеклянными бусами или кольцами Рашига, прибавляя в колбу, смотря по степени препарата чистоты (предварительная проба) 3–6 % от ее веса бихромата натрия и соответствующее количество серной кислоты. Иногда техническая валериановая кислота настолько загрязнена (т. е. содержит смолистые вещества), что ее сначала приходится просто перегонять без колонки, а затем уже подвергать обработке хромпиком и серной кислотой и одновременно ректификации.
Описанная очистка валериановой кислоты должна производиться на открытом воздухе под небольшой крышей, вследствие ее неприятного и чрезвычайно прилипчивого запаха.
6. Методы анализа РВОТНЫХ И ПРОТИВОРВОТНЫХ лекарственных СРЕДСТВ6.1 Методы анализа рвотных лекарственных средств. Анализ апоморфина
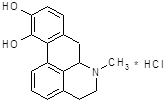
Описание. Белый, слегка сероватый или слегка желтоватый кристаллический порошок без запаха. На воздухе и на свету зеленеет [24].
Растворимость. Трудно растворим в воде и спирте, практически нерастворим в эфире и хлороформе.
Подлинность. При смачивании нескольких крупинок препарата 1 каплей азотной кислоты появляется кроваво-красное окрашивание
К 1 мл раствора препарата (1:100) прибавляют 1 мл 5% раствора гидрокарбоната натрия, 3 капли 0,1 н. раствора йода, 5 мл эфира и взбалтывают. Эфирный слой приобретает красно-фиолетовое окрашивание, а водный становится зеленым.
К 2 мл раствора препарата (1:100) прибавляют 0,5 мл разведенной азотной кислоты и фильтруют. К фильтрату прибавляют 0,5 мл раствора нитрата серебра; выделяется белый творожистый осадок.
Удельное вращение. От 46° до –52° (1,5 % раствор в 0,02 н. растворе соляной кислоты).
Цветность раствора. 0,1 г препарата растворяют в 10 мл воды. Свежеприготовленный раствор должен быть бесцветным.
Кислотность. рН 4,3–5,3 (1 % водный раствор, потенциометрически).
Продукты окисления. 5 мл эфира при встряхивании с 0,1 г препарата могут окрашиваться лишь вы слабо розовый цвет.
Морфин. 0,05 г препарата помещают на фильтр и промывают 3 мл разведенной соляной кислоты, предварительно охлажденной до 10°С. К фильтрату прибавляют 1 каплю реактива Майера; допускается лишь слабая опалесценция.
Потеря в весе при высушивании. Около 0,5 г препарата (точная навеска) сушат при 100–105°С до постоянного веса. Потеря в весе не должна превышать 4,5 %.
Сульфатная зола из 0,5 г препарата не должна превышать 0,1 %.
Количественное определение. Около 0,3 г препарата (точная навеска) растворяют в смеси 20 мл безводной уксусной кислоты и 10 мл раствора ацетата окисной ртути и титруют 0,1 н. раствором хлорной кислоты до зеленого окрашивания (индикатор кристаллический фиолетовый).
Параллельно проводят контрольный опыт.
1 мл 0,1 н. раствора хлорной кислоты соответствует 0,03038 г C17H17NO2 * HCl, которого в пересчете на сухое вещество должно быть не менее 98,0%
Хранение. Список А. В хорошо укупоренных банках оранжевого стекла.
6.2 Методы анализа противорвотных лекарственных средств
6.2.1 Анализ аминазина
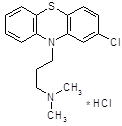
Описание. Белый или белый со слабым кремовым оттенком мелкокристаллический порошок. Слегка гигроскопичен, темнеет на свету [24].
Растворимость. Очень легко растворим в воде, легко растворим в 95% спирте и хлороформе, практически нерастворим в эфире и бензоле.
Подлинность. 0.05 г препарата растворяют в 10 мл воды, прибавляют 1 мл бромной воды и нагревают до кипения; получается прозрачный светло-малиновый раствор.
0,01 г препарата растворяют в 1 мл воды и прибавляют 2 капли концентрированной азотной кислоты. Раствор окрашивается в красный цвет и появляется белая муть. При прибавлении следующих 2–3 капель концентрированной азотной кислоты раствор становится прозрачным и бесцветным.
0,1 г препарата растворяют в 5 мл воды и прибавляют 0,5 мл раствора едкого натра, тотчас же выпадает осадок белого цвета; через 5 минут фильтруют через плотный бумажный фильтр. Фильтрат дает характерную реакцию на хлориды.
Температура плавления 194–198°.
Прозрачность и цветность раствора. 0,25 г препарата растворяют в 10 мл воды. Полученный раствор должен быть прозрачным и окраска его не должна быть интенсивнее эталона № 5а.
Кислотность. Раствор 0,5 г препарата в 10 мл свежепрокипяченной и охлажденной воды при добавлении 1 капли раствора метилового красного может окрашиваться в розовый цвет, переходящий от 0,05 мл 0,05 н. раствора едкого натра в оранжево-желтый.
Сульфаты. Раствор 0,2 г препарата в 10 мл воды должен выдерживать испытание на сульфаты (не более 0,05 % в препарате).
Хлорфенотиазин. 0,5 г препарата растирают в маленьком стакане с 10 мл бензола, фильтруют в делительную воронку и бензольный раствор промывают сначала 0,1 н. раствором соляной кислоты (3 раза по 2 мл), а затем водой (2 раза по 2 мл) Промытый бензольный слой фильтруют в фарфоровую чашку, выпаривают досуха на водяной бане, остаток переносят в пробирку 3 мл спирта, прибавляют 1 мл насыщенной на холоду бромной воды и нагревают на кипящей водяной бане около 2 минут до исчезновения желтой окраски от брома. Затем доводят раствор до 8 мл спиртом. Окраска полученного раствора не должна превышать окраски эталонного раствора.
Примечание. Приготовление эталонного раствора: 2 г гексагидрата хлорида кобальта (СоСl2•6Н2О) растворяют в 100 мл воды.
Органические примеси. 0,1 г препарата растворяют в 1 м спирта. 0,01 мл полученного раствора наносят на полоску быстрофильтрующей бумаги для хроматографии. Хроматографируют нисходящим методом в системе н-бутиловый спирт – вода – уксусная кислота (50:50:1) до тех пор, пока фронт растворителя не пройдет 12–15 см (примерно 5 часов). Подсушенную на воздухе хроматограмму опрыскивают реактивом Драгендорфа. Не должно быть пятна на линии старта.
Примечание. 1. Хроматограмму, проявленную реактивом Драгендорфа, водой не промывают. 2. Используют продольно разрезанную бумагу для хроматографии. Перед хроматографированием полоску с нанесенными веществами выдерживают в течение 30 минут в камере для насыщения.
Потеря в весе при высушивании. Около 0,5 г препарата (точная навеска) сушат при 100–105° до постоянного веса. Потеря в весе не должна превышать 0,5 %.
Сульфатная зола и тяжелые металлы. Сульфатная зола из 0,5 г препарата не должна превышать 0,1% и должна выдерживать испытание на тяжелые металлы (не более 0,001% в препарате).
Количественное определение. 0,25–0,30 г препарата (точная навеска) растворяют в смеси 30 мл ацетона и 5 мл раствора ацетата окисной ртути и прибавляют 1 мл насыщенного раствора метилового оранжевого в ацетоне. Титруют 0,1 н. раствором хлорной кислоты до розового окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,1 н. раствора хлорной кислоты соответствует 0,03553 г С17Н19СlN2S•НСl, которого в пересчете на сухое вещество должно быть не менее 99,0 % и не более 101,0 %.
Хранение. Список Б. В банках темного стекла, плотно закрытых пробками. залитыми парафином, в сухом, защищенном от света месте.
6.2.2 Анализ анестезина
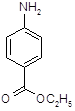
Описание. Белый кристаллический порошок без запаха, слабо горького вкуса. Вызывает на языке чувство онемения [24].
Растворимость. Очень мало растворим в воде, легко растворим в спирте, эфире, хлороформе, трудно растворим в жирных маслах и разведенной соляной кислоте.
Подлинность. Препарат дает характерную реакцию на ароматические первичные амины.
0,05 г препарата нагревают с 5 мл раствора едкого натра и приливают 0,1 н. раствор йода до неисчезающего желтого окрашивания; появляется запах йодоформа.
0,05 г препарата растворяют в 2 мл воды с 5 каплями разведенной соляной кислоты и прибавляют 2 мл раствора хлорамина. Через 2–3 минуты добавляют 2 мл эфира и взбалтывают; эфирный слой окрашивается в оранжевый цвет.
Температура плавления 89–91,5°.
Прозрачность и цветность раствора. Раствор 1 г препарата в 10 мл нейтрализованного по фенолфталеину спирта должен быть прозрачным и бесцветным.
Кислотность. К полученному раствору прибавляют 3 капли раствора фенолфталеина. Розовое окрашивание должно появиться от прибавления не более 0,1 мл 0,05 н. раствора едкого натра.
Хлориды. Раствор 1 г препарата в 10 мл спирта должен выдерживать испытание на хлориды (не более 0,002 % в препарате).
Органические примеси. 0,5 г препарата растворяют в 5 мл концентрированной серной кислоты. Окраска полученного раствора не должна быть интенсивнее эталона № 5а.
Сульфатная зола и тяжелые металлы. Сульфатная зола из 0,5 г препарата не должна превышать 0,1% и должна выдерживать испытание на тяжелые металлы (не более 0,001 % в препарате).
Количественное определение. Около 0,2 г препарата (точная навеска) растворяют в 10 мл воды и 10 мл разведенной соляной кислоты. Добавляют воды до общего объема 80 мл, 1 г бромида калия и при постоянном перемешивании титруют 0,1 мол раствором нитрита натрия, добавляя его в начале со скоростью 2 мл в минуту, а в конце титрования по 0,05 мл через минуту. В случае применения внутренних индикаторов используют нейтральный красный или тропеолин 00 в смеси с метиленовым синим.
1 мл 0,1 мол раствора нитрита натрия соответствует 0,01652 г С9Н11NО2, которого в препарате должно быть не менее 99,5 %.
Хранение. Список Б. В хорошо укупоренной таре, предохраняющей от действия света.
6.2.3 Анализ бромкамфоры
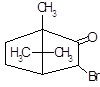
Описание. Бесцветные кристаллы или белый кристаллический порошок камфорного запаха и вкуса [24].
Растворимость. Очень мало растворим в воде, легко растворим в спирте, эфире, хлороформе и жирных маслах.
Подлинность. 0,1 г препарата растворяют в 3 мл спирта, прибавляют 1 мл раствора едкого натра и 0,3 г цинковой пыли. Смесь кипятят в течение 1-2 минут и после охлаждения фильтруют; фильтрат дает характерную реакцию А на бромиды.
Температура плавления 74–76°.
Кислотность или щелочность. 1 г препарата взбалтывают в течение 1 минуты с 20 мл свежепрокипяченной и охлажденной воды и фильтруют. К 5 мл этого фильтрата прибавляют 1 каплю раствора метилового красного; появившееся желтое окрашивание должно перейти в розовое от прибавления не более 0,05 мл 0,05 н. раствора соляной кислоты.
Галогены. 10 мл того же фильтрата должны выдерживать испытание на хлориды (не более 0,004 % в препарате).
Сульфатная зола из 0,5 г препарата не должна превышать 0,1 %.
Количественное определение. Около 2 г препарата (точная навеска) растворяют в 25 мл 95 % спирта в конической колбе емкостью 100 мл, прибавляют 10 мл свежеприготовленного 30 % раствора едкого кали, 2 капли раствора сульфата меди и 2 г цинковой пыли. Колбу соединяют с обратным холодильником и оставляют на 30–40 минут. Затем содержимое колбы кипятят на водяной бане в течение 30 минут. Кипячение прекращают, холодильник промывают 5 мл 95 % спирта и смесь снова кипятят в течение 5 минут.
После охлаждения жидкость сливают с осадка в мерную колбу емкостью 100 мл, осадок переносят на фильтр и промывают небольшими порциями воды в ту же мерную колбу до полного извлечения бромида и доводят водой до метки. Полученный раствор фильтруют через вату в сухую колбу. Первые 10–45 мл фильтрата отбрасывают. К 10 мл фильтрата прибавляют 5 мл разведенной азотной кислоты, 5 капель раствора железоаммониевых квасцов, 0,1 мл 0,1 н. раствора роданида аммония и титруют 0,1 н. раствором нитрата серебра до исчезновения красноватого окрашивания.
Из объема 0,1 н. раствора нитрата серебра, израсходованного на титрование, вычитают 0,1 мл (количество прибавленного 0,1 н. раствора роданида аммония) и полученную разность пересчитывают на бромкамфору.
1 мл 0,1 н. раствора нитрата серебра соответствует 0,02311 г С10Н15BrО, которой в препарате должно быть не менее 99,0 %.
Хранение. В хорошо укупоренных банках оранжевого стекла, в защищенном от света месте.
6.2.4 Анализ валидола
Описание. Прозрачная маслянистая бесцветная жидкость с запахом ментола [24].
Растворимость. Практически нерастворим в воде, очень легко растворим в спирте.
Подлинность. 1 г препарата растворяют в 1 мл концентрированной серной кислоты, прибавляют 1 мл раствора ванилина в серной кислоте, перемешивают и разбавляют 1 мл воды; появляется малиново-красное окрашивание.
Плотность 0,894–0,907.
Кислотность. 5 г препарата растворяют в 10 мл нейтрализованного по фенолфталеину спирта и прибавляют 1–2 капли раствора того же индикатора; розовое окрашивание должно появляться от прибавления не более 0,1 мл 0,05 н. раствора едкого натра.
Нелетучий остаток. 1 г препарата выпаривают на водяной бане до суха. Остаток не должен превышать 0,1 %.
Количественное определение. Около 2 г препарата (точная навеска) вносят в коническую колбу емкостью 250 мл, в которую предварительно помешают 20 мл бесцветного 1 н. спиртового раствора елкого кали. Кипятят на закрытой электроплитке с обратным холодильником в течение 5 часов; затем промывают холодильник и пробку 50 мл воды, охлаждают и титруют 0,5 н. раствором соляной кислоты до исчезновения розового окрашивания (индикатор – фенолфталеин).
Параллельно проводят контрольный опыт.
1 мл 0,5 н. раствора соляной кислоты соответствует 0,1202 г С15Н28О2, которого в препарате должно быть не менее 68,5 % и не более 75,0 %.
Хранение. В прохладном месте, в хорошо укупоренной таре.
7. СВОДНАЯ ТАБЛИЦА ПРЕПАРАТОВ
|
№ п/п |
Структурная формула |
Международное непатентованное название (синоним) |
Систематическое химическое название |
Методы получения |
Методы анализа |
Примечание |
7.1 Рвотные лекарственные препараты |
||||||
| 1. |
|
Апоморфина гидрохлорид 3,4-дигидрокси- апорфин |
[14], [16] | [24] | Белый, слегка сероватый или слегка желтоватый кристаллический порошок без запаха. Трудно растворим в воде (1:60) и спирте (1:50), практически не растворим в диэтиловом эфире и хлороформе. | |
| 2. |
|
Бромокриптина мезилат, Парлодел, Бромэргон, Правидел |
Бромокриптин 2-Бром-α-эргокриптин |
[17], [18] | ||
| 3. | NH4OH | Нашатырный спирт | Раствор аммиака 10% (Solutio Ammonii Caustici) | [24], [26] | Прозрачная, бесцветная жидкость. Растворима в воде и 96 % спирте. Относительная плотность 0, 892–0, 910. | |
| 4. |
|
Медный купорос, Медь сернокислая, | Меди сульфат | [24] | Синие кристаллы или синий кристаллический порошок без запаха, металлического вкуса. Легко растворим в воде (1:3 в холодной и 1:8 в кипящей). | |
| 5. |
|
Цинка сульфат | [24] | Бесцветные прозрачные кристаллы или мелкокристаллический порошок вяжущего вкуса, без запаха. Очень легко растворим в воде, нерастворим в спирте. | ||
7.2 Противорвотные лекарственные препараты |
||||||
| 1. |
|
Хлорпромазин, Ларгактил, Плегомазин |
Аминазин 2-Хлор-10-(3-диметиламино-пропил)-фенотиазина гидрохлорид |
[19], [20], [25] | [24], [26] | Белый или белый со слабым кремовым оттенком мелкокристаллический порошок. Слегка гигроскопичен, темнеет на свету. Очень легко растворим в воде, легко растворим в 95 % спирте и хлороформе, практически нерастворим в эфире и бензоле. Температура плавления 194–198°. |
| 2. |
|
Ампразин, Френил, Празин |
Пропазин 10-(3-Диметил-аминопропил)-фенотиазина гидрохлорид |
[24] | Белый или белый со слабым желтоватым оттенком кристаллический порошок. Очень легко растворим в воде. Гигроскопичен. На свету порошок и его растворы приобретают синевато-зеленую окраску. | |
| 3. |
|
Нозинан, Тизерцин |
Левомепромазин 2-Метокси-10-(3-диметиламино-2-метилпропил)-фенотиазина гидрохлорид |
|||
| 4. |
|
Терален |
Алимемазин 10-(3-Диметил-амино-2-метилпропил)-фенотиазин |
Белый или слегка желтоватый кристаллический порошок; растворим в воде и спирте. | ||
| 5. |
|
Хлорпипразин, Фентазин, Трелафон, Перфеназин |
Этаперазин 2-Хлор-10-{3-[1-(b-оксиэтил)- пиперазинил-4]-пропил)-фенотиазина дигидрохлорид |
[25] | Белый или белый с едва заметным сероватым оттенком кристаллический порошок. Легко растворим в воде, мало – в спирте. Гигроскопичен. Порошок и водные растворы разлагаются под действием света. Температура плавления 212–216°. | |
| 6. |
|
Метофеназат |
Френолон 3,4,5-Триметокси-бензоат 2-хлор-10-{3-[1-(ß- оксиэтил)-пиперазинил-4]-пропил)-фенотиазина дифумарат (или диэтансульфонат) |
|||
| 7. |
|
Стелазин |
Трифтазин 2-Трифторметил-10-[3-(1-метил- пиперазинил-4)-пропил]-фенотиазина дигидрохлорид |
[25] | [24] | Белый или слегка зеленовато-желтоватый кристаллический порошок без запаха. Легко растворим в воде, растворим в 95 % спирте, практически нерастворим в эфире и бензоле. На свету темнеет. Температура плавления 232–240°. |
| 8. |
|
Миренил, Модитен, Флуфеназин |
Фторфеназин 2-Трифторметил-10-{3-[1-(ß-оксиэтил)-пиперазинил-4]-пропил}-фенотиазина дигидрохлорид |
|||
| 9. |
|
Фторменазин-депо, Модитен-депо, Модекейт, Миренил-ретард |
Фторфеназин-деканоат 2-Трифторметил-10-{3-[1-(ß-каприноил- оксиэтил)-пиперазинил-4]-пропил}-фенотиазин |
|||
| 10. |
|
Мажептил |
Тиопроперазин 2-Диметил- сульфамидо-10-[3-(1-метил- пиперазинил-4)-пропил]-фенотиазин |
|||
| 11. |
|
Неулептил |
Перициазин 2-Циан-10-[3-(4-оксипиперидино)-пропил]-фенотиазин |
|||
| 12. |
|
Атозил, Фенерган, Пипольфен |
Дипразин 10-(2-Диметил-аминопропил)-фенотиазина гидрохлорид |
[24] | Белый кристаллический порошок. Очень легко растворим в воде, легко в спирте; рН 2,5 % водного раствора 3,9–4,9. Порошок и водные растворы темнеют под влиянием света. | |
| 13. |
|
Горестен, Трестен, Торекан |
Тиэтилперазин 2-(этилтио)-10-[3-(4-метилпиперазин-1-ил)пропил]-10H-фенотиазина дималеат |
|||
| 14. |
|
Хлотиксен, Минитиксен |
Хлорпротиксен Цис-2-Хлор-9-(3-диметиламинопропилиден)-тиоксантена гидрохлорид |
|||
| 15. |
|
Галофен |
Галоперидол 4-[4-(n-хлорфенил)-4-гидроксипиперидино]-41-фторбутиро- фенон |
[26] | Порошок белого или почти белого цвета. Практически не растворим в воде, мало растворим в 96 % спирте, метаноле и метиленхлориде. Температура плавления 150–153°. | |
| 16. |
|
Триседил |
Трифлуперидол 4-(мета-Трифторметилфенил)-1 [3'-(пара-фторбензоил)-пропил]пиперидинол- 4, или 4-[4-(мета-трифторметилфенил)-4-оксипиперидино]4'-фторбутирофенон |
|||
| 17. |
|
Дегидробензперидол, Дридол, Дролептан, Инапсин, Синтодрил |
Дроперидол 1-{1-[3-пара-Фторбензоил)пропил]-1,2,3,6-тетрагидро-4-пиридил}2-бензимидазолинон |
Светло-кремовый кристаллический порошок. При хранении на воздухе и на свету желтеет. Практически не растворим в воде, мало растворим в спирте. | ||
| 18. |
|
Догматил, Арминол, Конфидан, Эглонил |
Сульпирид N-(1-этил-2-пирролидинилметил)-2-метокси-5-сульфамоилбензамид |
|||
| 19. |
|
Мепробамат, Андаксин, Мильтаун |
Мепротан Дикарбамат 2-метил-2-пропилпропан-диола-1,3 |
Белый кристаллический порошок. Мало растворим в воде, легко – в спирте. | ||
| 20. |
|
Драмамин, Пермигал, Дедалон, Адразин, Авиомарин |
Дименгидринат 8-Хлортеофиллинат ß-диметиламино-этилового эфира бензгидрола |
|||
| 21. |
|
Бимарал, Клометол, Реглан, Церукал |
Метоклопрамид 4-Амино-5-хлор-N-(2-(диэтиламино)-этил)-2-метоксибензамида гидрохлорид |
|||
| 22. |
|
Навобан |
Трописетрон (1αН, 5αН)-тропан-3α-индолил-3-карбоксилат |
|||
| 23. |
|
Эметрон, Зофран |
Ондансетрон 2,3-дигидро-9-метил-3-((2-метил-1H-имидазол-1-ил)метил)-1H-карбазол-4(9H)-он |
|||
| 24. |
|
Китрил |
Гранисетрон 1-метил-N-(9-метил-9-аза-бицикло[3.3.1]нонан-3-ил)-1H-индол-3-карбоксамид |
|||
| 25. |
|
Бромурал, Аллувал, Дормиген, Изобромул, Сомнурол |
Бромизовал N-(α-Бромизо-валерианил)-мочевина |
Белый кристаллический порошок со слабым запахом, горьковатого вкуса. Очень мало растворим в воде, растворим в 95 % спирте. Температура плавления 145–150°. | ||
| 26. |
|
Клозапин, Алемоксан, Лепонекс |
Азалептин 8-Хлор -11-(4-метил-1-пиперазинил)-5Н-дибензо-[b,е][1,4]-диазепин |
Зеленовато-желтый мелкокристаллический порошок. Практически нерастворим в воде, трудно растворим в спирте. | ||
| 27. |
|
Скополамина гидробромид, Гиосцина гидробромид | Скополамин | [24] | Безцветные прозрачные кристаллы или белый кристаллический порошок. Легко растворим в воде (1:3), растворим в спирте (1:17). | |
| 28. |
|
Бензокаин |
Анестезин Этиловый эфир n-аминобензойной кислоты |
[21], [22] | [24] | Белый кристаллический порошок без запаха, слабо горького вкуса. Вызывает на языке чувство онемения. Очень мало растворим в воде, легко растворим в спирте, эфире, хлороформе, трудно растворим в жирных маслах и разведенной соляной кислоте. Температура плавления 89–91,5°. |
| 29. |
|
Дифенгидрамин гидрохлорид, Амидрил, Бенадрил, Рестамин |
Димедрол ß-Диметиламино-этилового эфира бензгидрола гидрохлорид |
[25] | [24] | Белый мелкокристаллический порошок, без запаха, горького вкуса; вызывает онемение языка. Гигроскопичен. Легко растворим в воде, очень легко – в спирте. Мало растворим в хлороформе, не растворим в эфире и бензоле. Температура плавления 166–170°. |
| 30. |
|
Меклозина гидрохлорид, Меклизин, Бонин |
Меклозин 1-[(4-хлорфенил)-метил]-4-[(3-метилфенил)-фенил]-пиперазин |
|||
| 31. |
|
Диметпрамид 5-нитро-4-диметиламино-2-метокси-N-(2-диэтиламиноэтил)-бензамида гидрохлорид |
||||
| 32. |
|
Бимарал, Албекс, Антемекс, Бромил, Дигезан, Эмеприд, Мепрамид |
Бромоприд 4-амино-5-бром-N-(2-N,N-диэтиламиноэтил)-2-метоксибензамид |
|||
| 33. |
|
Валидол Раствор ментола в ментиловом эфире изовалериановой кислоты |
[21] | [24] |
Прозрачная маслянистая бесцветная жидкость с запахом ментола. Плотность 0,894–0,907. Практически нерастворим в воде, очень легко растворим в спирте. |
|
| 34. |
|
Камфора бромистая | Бромкамфора | [14], [21] | [24] | Бесцветные кристаллы или белый кристаллический порошок слабого камфорного запаха и вкуса. Температура плавления 74–76°. Нерастворима в воде, легко растворим в эфире (1:2), спирте (1:9), хлороформе. |
| 35. |
|
Ментол | [24] |
Бесцветные кристаллы с запахом перечной мяты и холодящим вкусом. Не растворим в воде, растворяется в спирте, эфире, уксусной кислоте, жидком парафине, жирных маслах. При обыкновенной температуре летуч; перегоняется с водяным паром. При смещении с равным по весу количеством камфоры, хлоралгидрата, фенола, резорцина или тимола образует эвтектические смеси. Температура плавления 41–44°. |
||
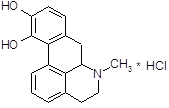
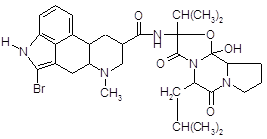
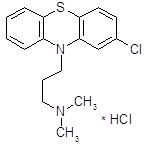
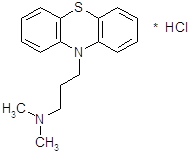
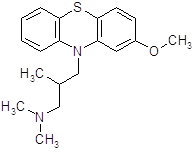
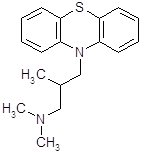
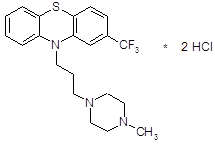

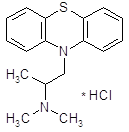
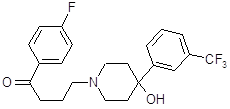
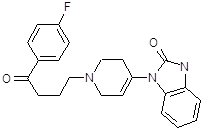
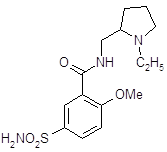
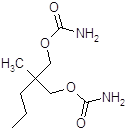
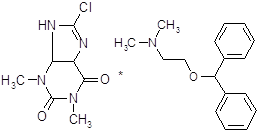
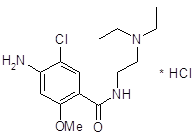
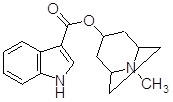
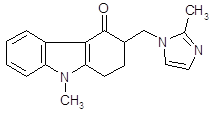
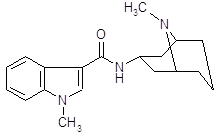
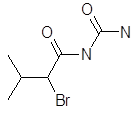
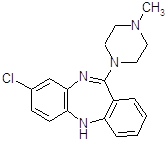
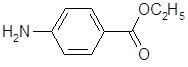
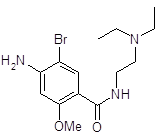
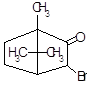
Словарь ТЕРМИНОВ
|
N. vagi (cardiaci superiores) – верхние сердечные ветви блуждающего нерва [27]. |
|
Аденогипофиз – доля гипофиза, имеющая железистое строение и функционирующая как железа внутренней секреции [27]. |
|
Алкалоз – форма нарушения кислотно-щелочного равновесия в организме, характеризующаяся сдвигом соотношения между анионами кислот и катионами оснований в сторону увеличения катионов [27]. |
|
Альдостерон – гормон животных и человека, вырабатываемый в коре надпочечников; регулирует минеральный обмен в организме, главным образом обмен Na+, К+ и воды [27]. |
|
Акинезия – отсутствие активных движений вследствие параличей, болей, неподвижности суставов и др. причин [27]. |
|
Амилоидоз – белково-углеводная дистрофия; образование и системное (или локальное) отложение в тканях белкового вещества амилоида; обусловлено генетически (например, при периодической болезни); может возникать как осложнение хронических инфекций (например, остеомиелита) и т. д. [27]. |
|
Анамнез (от греч. anamnesis – воспоминание) – совокупность сведений о развитии болезни, условиях жизни, перенесенных заболеваниях и др., собираемых с целью их использования для диагноза, прогноза, лечения, профилактики [27]. |
|
Анорексия (от греч. an – отрицание и orexis желание есть, аппетит) – отсутствие аппетита при наличии физиологической потребности в питании, обусловленное нарушениями деятельности пищевого центра [27]. |
|
Антральный (от греч. antron – пещера, полость тела) – относящийся к пещере, например сосцевидной, привратниковой [27]. |
| Афферентные волокна (от лат. afferens - приносящий) – несущие к органу или в него; передающие импульсы от рабочих органов (желез, мышц) к нервному центру [27]. |
|
Ахалазия пищевода (кардиоспазм) – стойкое функциональное нарушение проходимости нижнего (кардиального) отдела пищевода; проявляется дисфагией, загрудинной болью, срыгиванием [27]. |
|
Ацетонемия (кетонемия) – наличие в крови кетоновых тел; в норме 1,0 – 2,0 мг% (по ацетону) [27]. |
|
Ацетонурия (кетонурия) – повышенное выведение кетоновых тел с мочой; наблюдается при сахарном диабете, отравлениях ацетоном, голодании и т.д. [27]. |
|
Ацидурия (от лат. acidus – кислый и греч. uron – моча) – наследственная болезнь, обусловленная дефектом фермента метилмалонил-КоА-изомеразы-2 и проявляющаяся задержкой психического и физического развития [27]. |
|
Аэрофагия (от греч. phagein – есть, поглощать) – заглатывание избыточного количества воздуха с последующим его отрыгиванием; наблюдается при неправильном приеме пищи, при некоторых заболеваниях пищеварительного аппарата, при неврозах [27]. |
|
Булимия (греч. bulimia, буквально – бычий голод) – патологическое, резко усиленное чувство голода, сопровождающееся общей слабостью, болью в подложечной области; наблюдается при эндокринных и некоторых других заболеваниях [27]. |
|
Ваготомия (от лат. nervus vagus – блуждающий нерв и греч. tome – разрез) – хирургическая операция пересечения блуждающего нерва или его отдельных ветвей; применяется для лечения язвенной болезни [27]. |
|
Вагусным (вагальный) – связанный с блуждающим нервом [27]. |
|
Гастрит – воспалительное заболевание слизистой оболочки желудка. Причины – неправильное питание, воздействие алкоголя, никотина, пищевые отравления и др. Может быть острым (боли под ложечкой, тошнота, рвота) и хроническим (чувство тяжести и боли под ложечкой, изжога, отрыжка и др.) [27]. |
|
Гематоэнцефалический барьер (от греч. enkephalos – мозг) – физиологический механизм, регулирующий обмен веществ между кровью, спинномозговой жидкостью и мозгом. Защищает центральную нервную систему от проникновения чужеродных веществ, введенных в кровь, или продуктов нарушенного обмена веществ [27]. |
|
Гиперпаратиреоз – эндокринное заболевание, обусловленное избыточным образованием паратиреоидного гормона (преимущественно при аденоме околощитовидных желез): размягчение костей, патологические переломы, образование почечных камней, язв желудка и характеризующееся выраженным нарушением обмена кальция и фосфора [27]. |
|
Гиповолемия (от греч. hypo – уменьшение, франц. volume – объем и греч. haima – кровь; син.: олигемия) – уменьшенное общее количество крови [27]. |
|
Гипокалиемия – пониженное содержание калия в сыворотке крови [27]. |
|
Гипопаратиреоз (от лат. parathyreoidea околощитовидная железа) – синдром недостаточности функции околощитовидных желез, характеризующийся судорогами, нервными и психическими расстройствами, снижением содержания кальция в крови [27]. |
|
Глюкурониды – соединения глюкуроновой кислоты, образующиеся в организме при обезвреживании и выделении токсических веществ [27]. |
|
Депонирование (от лат. depono – складывать, откладывать) – 1) временное выключение каких-либо субстанций (например, форменных элементов крови, гормонов, жиров) из процессов циркуляции и обмена и их хранение в организме для последующего использования; системная реакция организма, служащая поддержанию гомеостаза; 2) накопление в тканях и органах лекарственных, радиоактивных, токсичных и других веществ, поступающих из окружающей среды [27]. |
|
Дефекация (от лат. defaecatio – очищение) рефлекторное выведение из пищеварительного тракта неусвоенных организмом остатков пищи; у млекопитающих животных и человека – опорожнение прямой кишки от кала. Центр рефлекса дефекации – в поясничной части спинного мозга [27]. |
|
Диафрагмальная грыжа – грыжа живота с выходом органов брюшной полости в грудную полость через расширенные щели или дефекты диафрагмы [27]. |
|
Дивертикулез тощей кишки наличие множественных дивертикулов (выпячиваний стенки полого органа, сообщающиеся с его полостью); наблюдается чаще в желудочно-кишечном тракте [27]. |
|
Дизентерия (от греч. enteron - кишка) острое инфекционное заболевание человека с поражением толстого кишечника (понос) и общей интоксикацией. Возбудители – бактерии рода шигелл. Заражение от больных и бактерионосителей через пищу, воду, грязные руки, мух [27]. |
|
Дизурия – общее название расстройств мочеиспускания, например, в виде его болезненности или затруднения выведения мочи из мочевого пузыря [27]. |
|
Дистальный (от лат. disto – отстою) расположенный дальше от срединной плоскости тела (в руке кисть – дистальный отдел) или от основного органа соответствующей системы (дистальные сосуды находятся дальше от сердца) [27]. |
|
Дисфагия (от греч. phagein – есть, глотать) расстройство глотания при заболеваниях глотки, пищевода, нервной системы [27]. |
|
Идиопатический (от греч. idios – сам по себе, отдельный и pathos – страдание, болезнь) – возникающий без видимых причин [27]. |
|
Интрамуральный (от лат. murus – стенка) внутристеночный, локализующийся в стенке полого органа или полости [27]. |
|
Катехоламины – производные пирокатехина; участвуют в обмене веществ и приспособительных реакциях организма, обеспечивая гомеостаз [27]. |
|
Клиренс (от англ. clearance – очищение) скорость очищения крови от какого-либо вещества в процессе его химических превращений [27]. |
|
Коклюш (франц. coqueluche) – острое инфекционное заболевание преимущественно детей (приступы судорожного кашля). Вызывается бактерией Борде-Жангу. Передается через воздух с капельками слизи [27]. |
|
Конъюгация (от лат. conjugatio – соединение) клеточный контакт микроорганизмов, во время которого происходит переход генетического материала из одной клетки в другую [27]. |
|
Культя – часть конечности или органа, оставшаяся после ампутации, или заменяющее конечность образование при ее врожденном недоразвитии. Культей называется также остаток нерва или бронха после их пересечения и перевязки и т. п. [27]. |
|
Лабиринтит – воспаление внутреннего уха чаще в результате перехода воспалительного процесса из среднего уха. Приводит к нарушению равновесия и к понижению или полной потере слуха [27]. |
|
Лимбическая система совокупность ряда структур головного мозга. Участвует в регуляции функций внутренних органов, обоняния, инстинктивного поведения, эмоций, памяти, сна, бодрствования и др. [27]. |
|
Менингит (от греч. meninx – мозговая оболочка) – гнойное или серьезное воспаление оболочек головного и спинного мозга, вызываемое бактериями, вирусами и др. Возникает как самостоятельное заболевание или как осложнение другого процесса. Проявляется головной болью, рвотой, расстройством сознания и т. д. [27]. |
|
Миопатия (от греч. myos – мышца и pathos страдание, болезнь) – общее название ряда наследственных болезней мышц, обусловленных нарушением сократительной способности мышечных волокон и проявляющихся мышечной слабостью, снижением тонуса, атрофией [27]. |
|
Никтурия (от греч. nyktos – ночь и uron моча) – выделение большей части суточного количества мочи ночью (нормальное соотношение количества ночной и дневной мочи 1:3). Может быть симптомом заболеваний почек (сочетается с полиурией) или сердечной недостаточности [27]. |
|
Паралич аккомодации нарушение зрительного восприятия [27]. |
|
Патогенез (от греч. pathos – страдание, болезнь) – механизмы развития заболеваний и патологических процессов (например, воспаления) [27]. |
|
Патофизиология (физиология патологическая) медицинская наука, изучающая общие закономерности возникновения, развития, течения и исхода болезней и патологических процессов [27]. |
|
Перистальтика (от греч. peristaltikos – обхватывающий и сжимающий), волнообразное сокращение стенок полых трубчатых органов (кишок, желудка, мочеточников и др.), способствующее продвижению их содержимого к выходным отверстиям [27]. |
|
Пилорический отдел (канал привратника) – конечная часть желудка, примыкающая к привратнику [27]. |
|
Пилоростеноз (от греч. pyloros – привратник и stenos – узкий) – сужение выходной части желудка – врожденное или приобретенное (рубец после химического ожога или язвы желудка). Основной симптом – рвота (при врожденном пилоростенозе – с двухнедельного возраста) [27]. |
|
Привратник – суженная часть желудка в месте его прохода в двенадцатиперстную кишку [27]. |
|
Продромальный период – стадия развития болезни, предшествующая ее основным клиническим проявлениям [27]. |
|
Проксимальный (от новолат. proximalis ближайший, от лат. proximus – ближний) – расположенный ближе к центру тела или к его медианной плоскости, например, плечо – проксимальный отдел по отношению к предплечью, а кисть – дистальный [27]. |
|
Реконвалесценция (позднелат. reconvalescentia) – период выздоровления после перенесенной болезни [27]. |
|
Резекция (от лат. resectio – отсечение) хирургическая операция: удаление части органа или анатомического образования, обычно с соединением его сохраненных частей [27]. |
|
Ретикулярная формация (formatio reticularis, син.: ретикулярная субстанция, сетевидная формация) – скопление нейронов, окруженное нервными волокнами, расположенное в центральных отделах ствола головного мозга и между боковыми и задними рогами спинного мозга; осуществляет активирующее воздействие на кору большого мозга и контролирует рефлекторную деятельность спинного мозга [27]. |
|
Рефлюкс (от лат. refluxus – обратное течение) – передвижение жидкого содержимого полых органов (пищеварительных, мочевыводящих и др.) в обратном направлении, например забрасывание желудочного содержимого в пищевод при диафрагмальной грыже [27]. |
|
Симпатическая нервная система часть вегетативной нервной системы, включающая нервные клетки грудного и верхнепоясничного отделов спинного мозга и нервные клетки пограничного симпатического ствола, солнечного сплетения, брыжеечных узлов, отростки которых иннервируют все органы. Симпатическая нервная система участвует в регуляции ряда функций организма: по ее волокнам проводятся импульсы, вызывающие повышение обмена веществ, учащение сердцебиений, сужение сосудов, расширение зрачков и др. [27]. |
|
Скарлатина (от лат. scarlatum – ярко-красный) инфекционное заболевание, преимущественно детей. Возбудитель гемолитический стрептококк. Характерны лихорадка, ангина, сыпь с последующим шелушением кожи. Заражение от больных и бактерионосителей через воздух [27]. |
|
Склеродермия – поражение кожи, характеризующееся ее диффузным или ограниченным уплотнением с последующим развитием фиброза и атрофии пораженных участков [27]. |
|
Соматотропин (от греч. somatos – тело и tropos направление) – гормон передней доли гипофиза, стимулирующий анаболические процессы [27]. |
|
Стереотипия – непроизвольное повторение однообразных, лишенных выразительности движений [27]. |
| Сфинктер (греч. sphinkter, от sphingo - сжимаю) – круговая мышца, суживающая или замыкающая при сокращении наружное (например, ротовое) или переходное (например, мочевого пузыря в мочеиспускательный канал) отверстие [27]. |
|
Тиреотоксический криз – внезапно возникающее, относительно кратковременное состояние у больного, характеризующееся появлением новых или усиленных симптомов, обусловленных повышенной функцией щитовидной железы [27]. |
|
Токсикоз (от греч. toxikon – яд) патологическое состояние, обусловленное действием на организм экзогенных токсинов (например, микробных) или вредных веществ эндогенного происхождения (например, при токсикозе беременных, тиреотоксикозе) [27]. |
|
Токсин – вещество бактериального, растительного или животного происхождения, способное при попадании в организм человека или животных вызывать заболевание или их гибель [27]. |
|
Уремия – патологическое состояние, обусловленное задержкой в крови азотистых шлаков, ацидозом и нарушениями электролитного, водного и осмотического равновесия при почечной недостаточности; обычно проявляется слабостью, апатией, ступором, гипотермией, артериальной гипертензией; может возникнуть кома [27]. |
|
Холецистит (от греч. chole желчь и kystis – пузырь) – острое или хроническое воспаление желчного пузыря, обычно при желчнокаменной болезни [27]. |
|
Цитостатические противоопухолевые препараты лекарственные средства, подавляющие деление клеток; используются главным образом для лечения злокачественных опухолей [27]. |
|
Эксикоз (обезвоживание организма) – болезненное состояние, обусловленное выделением значительного количества жидкости (полиурия, усиленное потоотделение, многократная рвота, понос и т. д.) при некоторых заболеваниях или водным голоданием. Проявления: жажда, потеря массы, в тяжелых случаях – судороги, психические нарушения, коллапс [27]. |
|
Экстрапирамидные расстройства нарушений координации движений с уменьшением их объема и дрожанием [27]. |
|
Эндогенный (от греч. endo – внутри и genes порождаемый) – возникающий в организме вследствие внутренних причин [27]. |
|
Энтеротоксин – общее название экзотоксинов, продуцируемых некоторыми видами бактерий (стафилоккоков, клостридий, эшерихий, шигелл) при их паразитировании в кишечнике [27]. |
|
Энтерохромаффиноподобные клетки – клетки эпителия кишечных крипт и слизистой оболочки желудка, содержащие в цитоплазме гранулы, способные восстанавливать серебро и хром из их смесей при отсутствии восстановителей. Вырабатывают биогенные амины (серотонин) [27]. |
|
Энцефалит – воспаление головного мозга (повышение температуры, головная боль, параличи, расстройства сознания и т. п.). Вызывается вирусами (первичный, эпидемический энцефалит) либо является осложнением других инфекционных заболеваний [27]. |
|
Эрготоксиновая группа (эрготоксин) смесь эргокристина, эргокорнина и эргокриптина [28]. |
|
Эфферентные волокна (от лат. efferens – выносящий), выносящие выводящие, передающие импульсы от нервных центров к рабочим органам, волокна [27]. |
1. Секреты гастроэнтерологии. – Под ред. Резник М. И., Новик Э. К./Электронная версия.
2. Справочник Харрисона по внутренним болезням. – Под ред. проф. В.П. Медведева/ Электронная версия.
3. Физиология человека. В 3-х томах. Т.3. Пер.с англ./Под ред. Р.Шмидта и Г. Тевса/ Электронная версия.
5. Cправочник по хирургии. – Под ред. С. Шварца, Дж. Шайерса, Ф. Спенсора/Электронная версия.
6. Справочник педиатра. – Под ред. О. П. Фомина/ Электронная версия.
7. Машковский М.Д. Лекарства ХХ века. – М.: ООО «Издательство Новая волна», 1998. – 320с.
8. Халецкий А.М. Фармацевтическая химия. – М.: Медицина, 1966. – 762 с.
9. Сергеев П.В., Шимановский Н.Л., Петров В.Н. Рецепторы физиологически активных веществ: Монография. – Волгоград: Издательство «Семь ветров», 1999. – 640 с.
11. Машковский М.Д. Лекарственные средства. В двух частях. Ч. 1. – 12-е изд., перераб. и доп. – М.: Медицина, 1998. – 736 с.
12. Кукес В.Г. Клиническая фармакология: Уч./Науч.ред. А.З. Байчурина. – 2 изд., перераб. и доп. – М.: ГЭОТАР Медицина, 1999. – 528 с.
13. Современные лекарственные препараты. – С. А. Крыжановский, М. Б. Вититнова. – М.: “РИПОЛ КЛАССИК”, 2000. – 1040 с.
14. Кацнельсон М.М. Приготовление синтетических химико-фармацевтических препаратов. 2-е изд. М.: Государственное техническое издательство, – 1923. – 291 с.
15. Майофис Л.С. Химия и технология химико-фармацевтических препаратов. – М.: Медицина, 1964. – 718с.
16. Синтез и фармакологическое изучение о, о´-диацилпроизводных апоморфина/ Буров Ю.В., Загоревский В.А., Варков А.И. и др.//Химико-фармацевтический журнал. 1985. – Вып. 10, № 19. – С. 1192–1195.
17. Выделение алкалоидов групп эрготоксина и эрготамина из отчественной спорыньи/ Тропп М.Я., Пелишенко И.И., Синилова Н.Г.// Медицинская промышленность СССР. – 1963. - № № 2. – С. 19–22.
19. Синтез 2-хлорфенотиазина/Русецкий Л.О.// Медицинская промышленность СССР. – 1962. - № 8. – С. 34–36.
20. К синтезу аминазина/Иванов П.Ю., Боканов А.И., Буданова Л.И., Кузовкин В.А. и др.//Химико-фармацевтический журнал. – 1986. – Вып. 21, № 9. – С. 1119–1121.
22. Абдуллаев М.Г/Получение анестезина гидрированием этилового эфира n-нитробензойной кислоты на палладиевых катализаторах//Химико-фармацевтический журнал. – 2001. – Вып. 35, 1. – С. 42–45.
23. Мелентьева Г.А., Антонова Л.А. Фармацевтическая химия. – М.: Медицина, 1985. – 480 с.
24. Государственная фармакопея СССР. 10-е издание. – М.: Медицина, 1968. – 1078 с.
25. Рубцов М.В., Байчиков А.Г. Синтетические химико-фармацевтические препараты. – М.: Медицина, 1971. 328 с.
26. Державна фармакопея України. 1-е видання. – Харків: РІРЕГ, 2001. – 556 с.
27. Энциклопедический словарь медицинских терминов/Электронная версия.
28. Химическая энциклопедия. В 5-ти томах. Т.5 – М.: «Большая российская энциклопедия», 1998. – 783 с.
© 2009 База Рефератов