
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Реферат: Биохимия сахарного диабета
Реферат: Биохимия сахарного диабета
Министерство здравоохранения Украины
Запорожский государственный медицинский университет
Кафедра биологической химии и лабораторной диагностики
Реферат
на тему:
«Биохимия сахарного диабета»
Выполнила:
студентка 2 курса 14 группы
медицинского факультета
Чмуль Карина Олеговна
г. Запорожье, 2007 г.
План
Ø Инсулинзависимый сахарный диабет
Ø Клеточный иммунитет
Ø Иммунный ответ на эндогенные и эндоцитированные белки
Ø Интерлейкин-1
Ø Модель аутоиммунной гибели -клеток
Ø дефицит инсулина
Ø Коматозные состояния (острые осложнения) при диабете
Ø Гликирование белков - одна из главных причин поздних осложнений сахарного диабета
Ø Диабетические ангиопатии
Ø Диабетические макроангиопатии.
Ø Микроангиопатии.
Инсулинзависимый сахарный диабет
1. При ИЗСД происходит разрушение -клеток в результате аутоиммунной реакции
Гипергликемия и другие первичные симптомы ИЗСД обусловлены дефицитом инсулина, который в свою очередь вызван уменьшением количества -клеток (а также островков Лангерганса) в поджелудочной железе. Множество экспериментальных и клинических исследований указывает на то, что разрушение островков происходит в результате клеточной аутоиммунной реакции.
При манифестации (т.е. первом клиническом проявлении) ИЗСД почти всегда обнаруживается воспалительная реакция в поджелудочной железе - инсулит. Панкреатический инфильтрат при ИЗСД содержит Т-лимфоциты, В-лимфоциты, натуральные киллеры и макрофаги. При этом инфильтрат образуется только в тех островках, в которых есть -клетки. В островках, продуцирующих глюкагон, соматостатин, но не содержащих -клеток, нет и инфильтрата. Такая локальность, точечность реакции указывает на то, что причиной ее являются компоненты и свойства, присущие только -клеткам. Как показывают многие наблюдения, специфичность повреждения -клеток может быть следствием клеточной аутоиммунной реакции.
Клеточный иммунитет. Основными молекулами, обеспечивающими клеточный иммунитет, являются Т-рецепторы и белки главного комплекса гистосовместимости (белки ГКГ). Эти два семейства молекул принадлежат к суперсемейству иммуноглобулинов, в которое входит также семейство иммуноглобулинов (антител), давших название всему суперсемейству. В отличие от антител, которые находятся в жидкостях организма в растворенном состоянии, Т-рецепторы и белки ГКГ - это интегральные белки клеточных мембран.
Т-рецепторы имеются на поверхности Т-лимфоцитов, а белки ГКГ - на поверхности практически всех клеток. Т-рецепторы представляют собой гетеродимеры , содержащие межцепочечную дисульфидную связь. Каждая цепь содержит глобулярные вариабильный и константный домены, экспонированные на наружной поверхности мембраны, а также трансмембранный домен и короткий цитоплазматический домен:
|
Строение Т-рецепторов (а) и белков ГКГ классов I (б) и II (в). Стрелки указывают на пептиды - лиганды белков ГКГ; М - 2-микроглобулин |
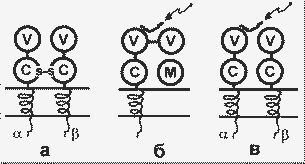
Т-рецептор составляет часть многомолекулярного белкового комплекса, включающего в общей сложности 7- 9 пронизывающих мембрану пептидных цепей. Этот комплекс формируется в цитозоле и затем включается в мембрану. Существует множество клонов Т-лимфоцитов, различающихся по структуре вариабильного домена, т.е. множество Т-рецепторов с разной специфичностью к лигандам. Разнообразие Т-рецепторов возникает так же, как и разнообразие антител, т.е. в результате соматической рекомбинации генов. Лигандами для Т-рецепторов служат короткие пептиды (10 - 20 аминокислотных остатков), которые образуются из чужеродных белков в результате протеолитической фрагментации. При этом для узнавания рецепторами необходимо, чтобы такие пептиды были соединены с белками ГКГ.
Известны два класса белков ГКГ, несколько различающихся по структуре и функциям. Белки класса I содержат две нековалентно связанные пептидные цепи - легкую и тяжелую. Тяжелая цепь своей большой N-концевой частью экспонирована на наружной поверхности клеточной мембраны, далее следуют небольшие трансмембранный и цитоплазматический домены. Легкая цепь представлена 2-микроглобулином (2m). Внеклеточная часть тяжелой цепи содержит три глобулярных домена: 1 и 2 - вариабильные домены, 3 - константный домен, сходный по структуре с пептидом 2m.
Белки ГКГ класса II - это гомодимеры; на поверхности клетки экспонированы вариабельный и константный глобулярные домены обеих цепей.
Белки ГКГ класса I имеются практически во всех клетках организма человека, а класса II - только в макрофагах, В-лимфоцитах и некоторых специализированных эпителиальных клетках. В геноме человека имеется лишь несколько генов (генных локусов) белков ГКГ (гены HLA). Однако в популяциях человека известно большое количество аллельных вариантов этих белков - варианты белков класса I и варианты белков класса II; отдельные индивиды могут наследовать лишь один (гомозиготы) или два (гетерозиготы) из этих вариантов, причем вероятность наследования разными индивидами одинаковых вариантов ничтожна. Т.о. между людьми существуют индивидуальные различия по белкам ГКГ. Именно с этим связана трансплантационная несовместимость индивидов.
Белки ГКГ являются рецепторами небольших пептидов (длиной в 10 - 20 аминокислотных остатков). Центр связывания этих пептидов образуют вариабельные домены белков ГКГ. Пептиды-лиганды могут образоваться в результате протеолитической фрагментации как собственных белков организма, так и чужеродных белков; в последнем случае пептиды-лиганды служат антигенами, вызывают иммунную реакцию с участием Т-лимфоцитов. К пептидам, образовавшимся из собственных нормальных (не мутантных) белков на ранних стадиях эмбрионального развития вырабатывается иммунологическая толерантность.
Комплекс белка ГКГ с пептидом служит лигандом Т-рецептора определенного клона Т-лимфоцитов. Т-лимфоцит своим Т-рецептором присоединяется к клетке, представившей на своей поверхности комплекс ГКГ/пептид, и если пептид в этом комплексе происходит не из собственного, а из чужеродного белка, Т-лимфоцит активируется, и включается механизм уничтожения клетки, несущей чужеродный пептид. Подчеркнем, что Т-рецептор связан не отдельно с белком ГКГ, и не отдельно с петидом-антигеном, а именно с комплексом этих молекул, которые вместе и в равной мере участвуют в образовании центра связывания для Т-рецепторов. Т.о. специфичность иммунного ответа есть результат вариабельности белков ГКГ, которые определяют и выбор пептида-антигена, и выбор Т-лимфоцита соответствующего клона.
Т-лимфоциты в организме человека представлены тремя типами: цитотоксические Т-лимфоциты (Т-киллеры), имеющие механизм уничтожения клеток, и два типа лимфоцитов, выполняющих регуляторные функции - Т-хелперы и Т-супрессоры. Т-хелперы, присоединившие антиген, стимулируют остальные компоненты иммунной системы: специфичные к данному антигену другие Т-лимфоциты, а также и В-лимфоциты. Т-супрессоры, наоборот, подавляют активность этих клеток. Т-хелперы, вероятно, играют главную роль в инициации иммунного ответа. В частности, пролиферация и окончательная дифференцировка В-лимфоцитов, узнавших чужеродный антиген, требует активации Т-лимфоцитами.
Таблица 2. Иммунный ответ на эндогенные и эндоцитированные белки
|
|

Чужеродные белки могут появиться в клетке двумя путями: 1) образоваться в самой клетке (вирусные белки, мутантные белки); 2) проникнуть путем эндоцитоза в клетки макрофагов и некоторых других фагоцитирующих клеток (любые белки, появляющиеся в жидкостях организма). Ответ клеточного иммунитета в этих случаях будет несколько различным (табл. 2).
На рисунке приведена схема инициации клеточного иммунного ответа на эндоцитированный чужеродный белок:
|
Инициация клеточного иммунного ответа |
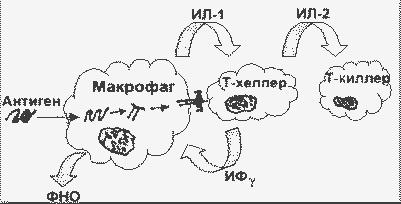
Антиген (Аг), обычно растворимый белок , часто гликопротеин, эндоцитируется антигенпредставляющими клетками (АПК; например, тканевыми макрофагами или В-лимфоцитами). В эндоцитозе участвует рецептор антигена на поверхности АПК. Комплекс Аг-рецептор интернализуется, в эндосоме происходит частичный протеолиз с образованием пептидов длиной в 10 - 20 аминокислотных остатков, пептиды соединяются с белками класса II главного комплекса гистосовместимости. Затем эндосома сливается с плазматической мембраной, и комплекс антигенный пептид/класс II-ГКГ экспонируется на поверхности клетки. Экспонированный комплекс может быть распознан Т-хелперами специфического клона, несущими подходящий Т-рецептор.
Когда Аг узнается Т-хелпером, он (Т-хелпер) активируется прежде всего в отношении транскрипции ряда цитокиновых генов. Продукция цитокинов (см. ниже) вызывает хемотаксис лейкоцитов к месту, где происходят эти события, активацию эндотелиальных клеток, пролиферацию и дифференцировку рекрутированных лейкоцитов, апоптоз и много других биологических активностей.
2. Интерлейкин-1 может быть токсичным для -клеток
В развитии клеточной аутоиммунной реакции участвуют цитокины. Это сигнальные молекулы паракринного и аутокринного действия, но некоторые из них иногда бывают и в крови в физиологически активной концентрации. Известны десятки разных цитокинов. К ним относятся интерлейкины (лимфокины и монокины), интерфероны, пептидные факторы роста, колониестимулирующие факторы. Цитокины представляют собой гликопротеины, содержащие 100 - 200 аминокислотных остатков. Большинство цитокинов образуется и действует во многих типах клеток и реагирует на разные стимулы, включая механическое повреждение, вирусную инфекцию, метаболические нарушения и др. Исключение составляют интерлейкины (ИЛ-1 и ИЛ-1) - их синтез регулируется специфическими сигналами и в небольшом количестве типов клеток.
Цитокины содержатся в тканях в пикомолярных и наномолярных концентрациях, и с высоким сродством взаимодействуют со специфическими рецепторами на наружной поверхности плазматической мембраны клеток. Цитокины участвуют в регуляции пролиферации, дифференцировки, хемотаксиса, секреции, апоптоза. ИЛ-1, фактор некроза опухолей (ФНО) и интерферон (ИФg) являются главными медиаторами развития острой фазы воспаления, инфекции и травмы. Они имеют перекрывающуюся, но все же разную биологическую активность. Клетки разных типов, или разной степени дифференцированности, или находящиеся в разном функциональном состоянии могут по-разному реагировать на один и тот же цитокин.
Цитокины действуют на клетки через специфические мембранные рецепторы и протеинкиназные каскады, в результате активируется фактор транскрипции - белок, который транслоцируется в ядро клетки, находит специфическую последовательность ДНК в промоторе гена, являющегося мишенью данного цитокина, и активирует этот ген.
В экспериментах с изолированными островками Лангерганса животных показано, что ИЛ-1 практически полностью подавляет стимулированную глюкозой секрецию инсулина и нарушает нормальную структуру островков. В островках снижается выживаемость клеток, отмечается фрагментация ДНК, уменьшается содержание ДНК, т.е. индуцируется апоптоз. При этом повреждаются преимущественно -клетки; вероятно, это связано с тем, что в островках именно -клетки имеют наибольшую плотность рецепторов ИЛ-1. Глюкоза защищает клетки от токсического действия ИЛ-1 (увеличивает выживаемость клеток). При этом индуцируется синтез белков, в частности bcl-2, ингибирующего апоптоз.
Цитокины ИФg и ФНОa усиливают токсическое действие ИЛ-1: в их присутствии ИЛ-1 токсичен для островков в гораздо меньших концентрациях. Другие цитокины не проявляют токсического действия в отношении островков.
ИЛ-1 индуцирует, в частности, синтез фермента NO-синтазы.Оксид азота NO - короткоживущий свободный радикал с высокой реакционной способностью. Он участвует в регуляции ряда физиологических функций, например, регулирует тонус сосудов (сосудорасширяющее действие), обладает противоопухолевым действием, токсичен для микроорганизмов. NO образуется при действии NO-синтазы (NOS), превращающей аргинин и кислород в цитруллин и NO. Есть два основных типа NO-синтазы: конститутивная форма (обнаружена в основном в нейронах и эндотелиальных клетках) и индуцибельная форма (iNOS) (во многих клетках, в том числе в b-клетках островков). Синтез iNOS индуцируется цитокинами и бактериальными липополисахаридами; фермент продуцирует значительно больше NO, чем конститутивные формы. Повидимому, iNOS и NO служат одним из иеханизмов защиты от микроорганизмов. NO проявляет летальное действие по отношению к простейшим, грибкам, бактериям и вирусам.
В островках Лангерганса iNOS образуется, по-видимому, только в b-клетках. В островках человека синтез мРНК и белка iNOS индуцируется при одновременном наличии двух или трех цитокинов: ИЛ-1 + ИФg или ИЛ-1 + ИФg + ФНО. В целом повреждение и гибель -клеток при аутоиммунной реакции можно представить следующим образом:
|
Модель аутоиммунной гибели -клеток |
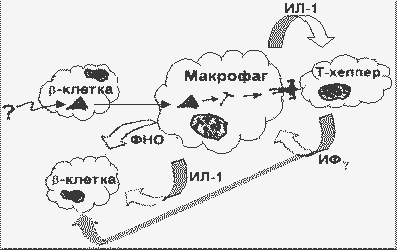
В ранней фазе иммунного ответа происходит взаимодействие одной АПК с одной Аг-узнающей клеткой. При этом повышается локальная концентрация цитокинов с паракринным действием на ближайшее окружение. Позднее развивается воспалительная реакция с участием активных иммунокомпетентных клеток, происходит секреция цитокинов, активация протеаз, образование кислородных радикалов, других иммунных медиаторов. Т.о. гибель клеток происходит, по-видимому, как по механизму некроза (воспаление), так и по механизму апоптоза.
Интерферон g (ИФg) обеспечивает положительную обратную связь с макрофагами в отношении продукции ИЛ-1 и ФНО, вследствие чего начавшийся с одной клетки иммунный ответ не затухает, а амплифицируется.
Остается неясным вопрос о природе антигена, запускающего реакцию клеточного иммунитета, избирательно направленную на -клетки. Интересные результаты получены в исследованиях на мышах линии NOD (non obesity diabetes) с высокой генетической предрасположенностью к ИЗСД. Из тканей этих мышей выделены клоны лимфоцитов, введение которых здоровым мышам вызывает диабет. Кроме того, такие лимфоциты оказались способными связывать инсулин, причем узнаваемой частью почти всегда был фрагмент -цепи, включающий 9 - 23 аминокислотные остатки (пептид В). В этих лимфоцитах пептид В соединен с белками ГКГ класса II. Авторы допускают, что инсулин может быть первичным аутоантигеном при ИЗСД у мышей NOD, а возможно и у человека.
Аутоиммунный процесс в островках поджелудочной железы развивается в течение нескольких лет и приводит к гибели основной массы (около 80%) -клеток до клинического дебюта болезни. В результате дефицита инсулина нарушается складирование энергоносителей и проявляется клиническая картина ИЗСД.
3. При дефиците инсулина нарушается синтез гликогена и жиров
При сахарном диабете инсулин-глюкагоновый индекс снижен. Это связано не только с уменьшением секреции инсулина, но и с увеличением секреции глюкагона (инсулин ингибирует секрецию глюкагона). В результате ослаблена стимуляция процессов складирования и усилена стимуляция мобилизации запасов, усилена настолько, что печень, мышцы, жировая ткань даже после приема пищи функционируют в режиме постабсорбтивного состояния. В этой драматической коллизии продукты переваривания, а также их метаболиты, вместо того, чтобы складироваться в форме гликогена и жиров, циркулируют в крови. Вероятно, в какой-то мере происходят и затратные циклические процессы типа одновременно протекающих гликолиза и глюконеогенеза, или синтеза и распада жиров и т.п..
Для всех форм сахарного диабета характерна сниженная толерантность к глюкозе, т.е. гиперглюкоземия после приема пищи или даже и натощак. При концентрации глюкозы в крови больше 180 мг/дл наступает глюкозурия. Повышена концентрация в крови липопротеинов (главным образом ЛОНП), свободных жирных кислот, кетоновых тел. В свою очередь гиперглюкоземия является основной причиной как острых, так и поздних осложнений диабета.
4. Коматозные состояния (острые осложнения) при диабете развиваются в результате нарушения обмена глюкозы и жиров
Коматозные состояния при сахарном диабете могут быть разного патогенеза. Различают три основные формы:
1. кетоацидотическая кома, с абсолютной инсулиновой недостаточностью
2. гиперосмолярная кома, с умеренной недостаточностью инсулина
3. лактатацидотическая кома, с выраженной гипоксией, сепсисом, сердечно-сосудистым шоком.
Кроме того, при инсулинотерапии сахарного диабета может быть гипогликемическая кома, связанная с передозировкой инсулина. Первые три состояния могут развиваться не только при сахарном диабете, но и при действии многих других факторов (токсических, инфекционных и др.).
Три основные формы коматозного состояния практически никогда не встречаются в чистом виде, однако обычно преобладают проявления какой-нибудь одной из форм (часто - гиперосмолярной), что и дает основания для выделения основных форм.
Первичной причиной кетоацидоза является инсулиновая недостаточность: в период комы С-пептид и иммунореактивный инсулин (ИРИ) в крови не определяются. Гипергликемия отмечается всегда, 20 - 30 ммоль/л, а иногда и более. Ацидоз при диабетической коме является следствием накопления органических кислот - кетоновых тел, а также лактата и пирувата. Концентрация кетоновых тел достигает 2 ммоль/дл (в 200 раз больше нормы); она повышается не только вследствие синтеза в печени, но и потому, что снижается экскреция кетоновых тел в связи с олигурией и анурией, которая часто бывает при коме. Снижение рН крови наблюдается всегда, до 7 и ниже (норма 7,4).
Развивается дегидратация, дефицит воды может быть до 10% от общей массы тела. Количество циркулирующей жидкости уменьшается на 25 - 30%, в результате снижается кровяное давление.
Кислородное и энергетическое голодание миокарда, уменьшение объема крови ведут к сердечно-сосудистой недостаточности. Могут быть повышение свертываемости крови, инфаркт миокарда, инфаркты паренхиматозных органов, инсульт, периферические тромбозы.
Диабетическая кома развивается медленно, в течение нескольких дней, но иногда может развиться за несколько часов. Появляются тошнота, рвота, черты лица заостряются, глаза западают, нарастает безучастность к окружающему, заторможенность, переходящая в глубокую кому (полностью выключенное сознание, отсутствие рефлексов, атония мышц и др.). В помещении, где находится больной, ощущается явственный запах ацетона. Артериальное давление снижено, почти всегда наблюдается олигурия или анурия.
Диабетическая кома требует немедленного лечения, которое включает следующие мероприятия:
1. ликвидация инсулиновой недостаточности путем введения инсулина в дозах, обеспечивающих постепенное снижение концентрации глюкозы в крови до уровня, близкого к нормальному
2. регидратацию организма путем введения жидкости
3. восстановление нормального солевого состава и рН жидкостей организма путем введения соответствующих солевых растворов
4. восстановление запасов гликогена в организме.
Проявления комы обычно ликвидируются в течение 2 - 3 дней при непрерывно продолжающемся лечении, причем лечение в первые несколько часов имеет решающее значение для спасения жизни больного.
До развития методов лечения диабета инсулином больные умирали вскоре после начала болезни от диабетической комы. Но и теперь кома наблюдается нередко. В частности, первое проявление болезни в 15 - 30% случаев сопровождается кетоацидозом и комой. Смертность от диабетической комы остается высокой - от 1 до 30%. Но основной причиной смерти больных диабетом в настоящее время являются поздние осложнения.
5. Гликирование белков - одна из главных причин поздних осложнений сахарного диабета
Поздние осложнения сахарного диабета связаны прежде всего с повреждением кровеносных сосудов (диабетические ангиопатии). Основной механизм повреждения тканей - гликирование (гликозилирование) белков, неферментативная реакция глюкозы со свободными аминогруппами белковой молекулы (Лиз, Арг, N-концевая аминокислота):
|
|
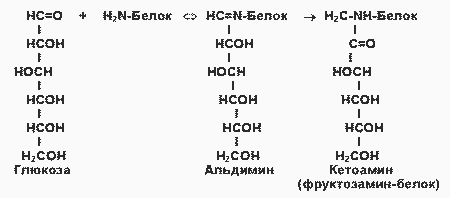
При этом образуется нестабильная альдиминовая группировка, которая может превращаться в ряд других, более стабильных соединений (“ранние продукты гликозилирования”). Понятно, что при этом функции белка могут быть нарушены в результате изменения заряда белковой молекулы, ее конформации или блокирования активного центра. Глюкозилирование - медленная реакция, в тканях здоровых людей обнаруживаются лишь небольшие количества гликозилированных белков. При гипергликемии реакция существенно ускоряется. Например, у больных диабетом в состоянии гипергликемии содержание одного из гликозилированных гемоглобинов - HBA1c - в течение 2 -3 недель увеличивается в 2 - 3 раза. Степень гликозилирования разных белков неодинакова; в основном она зависит от скорости обновления данного белка. В медленно обменивающихся белках накапливается больше модифицированных аминогрупп. Кроме того, в таких белках происходят дальнейшие изменения углеводных остатков - перестройки структуры, окислительные превращения, в результате которых образуются разнообразные “поздние продукты гликозилирования (ППГ), часто коричневого цвета, флуоресцирующие, и некоторые из них обладают высокой реакционной активностью и способностью дополнительно повреждать белки, в т. ч. образовывать поперечные сшивки между молекулами белков. К медленно обменивающимся белкам относятся многие белки соединительно-тканных образований, межклеточного матрикса, базальных мембран. К тому же белки этих структур непосредственно контактируют с межклеточной жидкостью, в которой концентрация глюкозы такая же, как в крови (в клетках она обычно гораздо ниже в результате использования глюкозы в метаболических процессах). В этих структурах ППГ накапливается с возрастом, и накопление сильно ускоряется при сахарном диабете.
ППГ-белки могут гидролизоваться макрофагами (с участием ППГ-рецепторов) или межклеточными протеолитическими системами с образованием ППГ-пептидов, часто длиной около 30 аминокислотных остатков. ППГ-белки, и особенно образующиеся в результате их гидролиза ППГ-пептиды, попадают и в кровоток. Концентрация ППГ-пептидов в крови резко повышается при почечной недостаточности разного происхождения, в том числе при диабетической нефропатии. Это связано с тем, что элиминация ППГ-пептидов поисходит с участием почек: ППГ-пептиды фильтруются в клубочках, реабсорбируются клетками проксимальных канальцев и катаболизируются в лизосомах этих клеток.
В экспериментах на крысах показано, что введение ППГ-белков в кровь приводит к ковалентному связыванию этих белков с белками межклеточного матрикса во многих тканях и к появлению структурных и функциональных нарушений, сходных с теми, которые бывают при сахарном диабете.
ППГ проявляют многообразную биологическую активность: повышают проницаемость эндотелиальных клеток, соединяются с рецепторами макрофагов, эндотелиальных и мезангиальных клеток, активируют макрофаги к секреции цитокинов (рецепторным путем), подавляют образование NO и соответственно ингибируют расширение сосудов, усиливают окисление ЛНП. В крови больных диабетом обнаруживаются антитела к ППГ-пептидам.
6. Диабетические ангиопатии
Первичные проявления ангиопатий связаны с повреждением базальных мембран сосудов. Базальные мембраны (БМ) представляют собой пленки, на которых “растут” все клетки организма, кроме клеток соединительной ткани и крови: по одну сторону располагается клетка или слой клеток, а другой стороной БМ контактирует с фиброретикулярным межклеточным матриксом:
|
Базальные мембраны разных органов (А) и капилляров почечного клубочка (Б): а – люмен капилляры; б – полость Боуменовой капсулы; 1 – эндотелий; 2,3,4 – БМ клубочка (2 – lamina rara interna, 3 – lamina densa, 4 – lamina rara externa); 5 – подоцит, отростками контактирующий с БМ. |
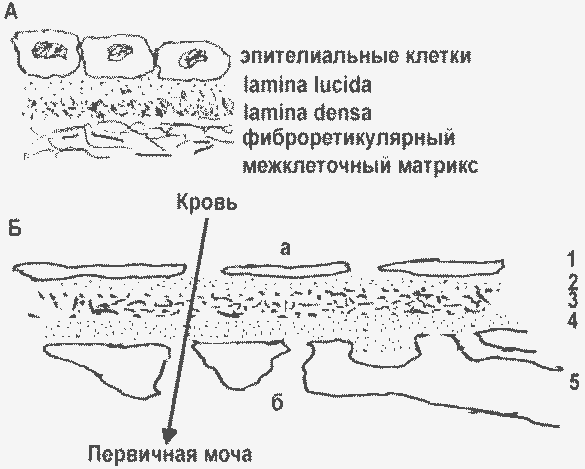
Эндотелий кровеносных сосудов, в том числе капилляров, тоже располагается на базальных мембранах. В отличие от всех прочих органов, в капиллярах почечного клубочка БМ трехслойна, а клетки располагаются по обе ее стороны.
В построении БМ участвуют коллагены, протеогликаны, неколлагеновые гликопротеины. Все компоненты БМ синтезируются прилегающими к ним клетками. Специальные функции выполняют интегрины - белки плазматической мембраны клеток, соединяющие клетку с БМ.
Коллаген IV типа - основной структурный белок базальных мембран. В геноме человека имеется шесть локусов, кодирующих шесть различающихся пептидных цепей, из которых строятся трехцепочечные молекулы коллагена IV. Чаще всего коллаген IV содержит цепи 1(IV) и 2(IV) в составе гетеротримеров [1(IV)]22(IV). Коллаген IV относится к сетеобразующим коллагенам. Взаимодействуя С-концевыми глобулярными доменами, молекулы образуют димеры, а при взаимодействии N-концевыми глобулярными доменами - тетрамеры.
Наряду с этими взаимодействиям конец в конец возможны и латеральные взаимодействия трехцепочечных спиральных доменов, в том числе с образованием суперспиралей. В конечном счете образуется сетевидная трехмерная структура с гексагональными ячейками размером 170 нм. Коллаген IV имеет также центры связывания с некоторыми белками клеточной мембраны, в том числе с интегринами.
Значительную часть массы БМ составляют протеогликаны. Эти молекулы содержат белковое ядро и ковалентно связанные с ним гликозамингликаны. В БМ в наибольших количествах содержатся гепарансульфат-протеогликаны (ГСПГ), и в значительно меньших - хондроитинсульфат-протеогликаны.
Гепарансульфат представляет собой неразветвленную цепь, построенную из глюкуроновой кислоты и глюкозамина, с последовательностью (ГлкА-ГлкN)n. Остаток глюкозамина может быть сульфирован по 2-й, 3-й и 6-й позициям. Молекулярная масса цепей обычно от 50 до 100 кДа. С одним белковым ядром ГСПГ обычно связано несколько цепей гепарансульфата. ГСПГ в БМ соединяется с коллагеном IV и ламинином определенными центрами белкового ядра и цепей гепарансульфата. Кроме того, ГСПГ разными способами может быть связан с поверхностью клеток, с участием как гликозамингликановой части, так и белкового ядра.
Ламинин - специфичный для БМ неколлагеновый гликопротеин. Молекула ламинина - тример , имеет крестообразную форму, с тремя короткими ветвями и одной длинной:
|
Молекулы базальных мембран. А. Многомолекулярные структуры, образуемые коллагеном IV: 1 - тетрамер; 2 - димер; 3 - мономер. Б. Фибронектин. В. Основные гепарансульфат-протеогликаны БМК: 1 - перлекан; 2 - ГСПГ 200 кДа; 3 - ГСПГ 350 кДа (горизонтальные линии - пептидные цепи, вертикальные линии - гепарансульфатные цепи). Г. Ламинин. |
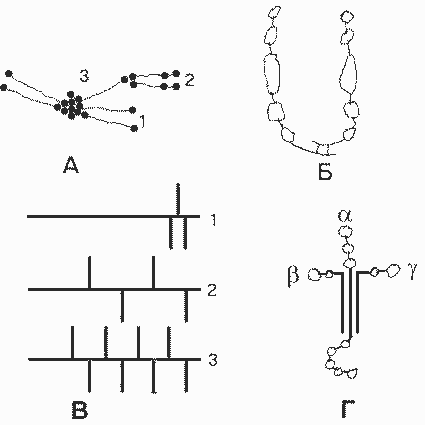
Известны три разные цепи , три цепи и две цепи . Варианты цепей могут комбинироваться по-разному при образовании тримерной молекулы. Пока обнаружено семь разных ламининов. Каждая из ветвей содержит глобулярные домены, которые имеют ряд центров свяэывания разных лигандов. Короткие ветви участвуют в образовании БМ путем взаимодейсв с другими молекулами ламинина, с коллагеном IV (при участии еще одного белка - нидогена), а также с интегрином 11 клеточной мембраны. Глобулярный домен длинной ветви участвует в межклеточной адгезии, взаимодействуя с разными интегринами и другими белками плазматической мембраны клеток. Длинная ветвь взаимодействует также с гепарансульфатными протеогликанами.
Интегрины представляют собой трансмембранные гликопротеины, -димеры. Каждая цепь пересекает мембрану один раз. Обе цепи имеют большие внеклеточные домены (аминоконцевые), образующие центр связывания, комплементарный соответствующему лиганду - компоненту матрикса. Внутриклеточный домен взаимодействуют с актиновым цитоскелетом при посредстве ряда промежуточных белков. С местом взаимодействия интегринов с цитоскелетом соседствуют сигнальные белки, которые активируются, когда к внеклеточному домену интегрина присоединяется лиганд. Таким лигандом могут быть ламинины, коллаген IV, протеогликаны.
Сила, с которой интегрин связывается с лигандом, может быстро и с высокой точностью регулироваться - свойство, которое называют “модуляция сродства”. В покоящемся (“неактивном”) состоянии интегрины имеют низкое сродство к своим лигандам, и это характерно для нормальных физиологических условий. Определенные стимулы превращают их в активные рецепторы с высоким сродством к лигандам. Это позволяет клеткам быстро приспосабливать их адгезивные свойства к изменившимся условиям без изменения количества молекул адгезии. А поскольку сигнал, полученный интегрином, передается в клетку, то могут изменяться не только адгезивные свойства, но и внутриклеточные процессы.
Благодаря этим свойствам интегрины оказываются участниками многих фундаментальных физиологических и патофизиологических процессов, включая эмбриогенез, морфогенез, заживление ран, воспаление, миграцию опухолевых клеток, миграцию лейкоцитов.
Таким образом, молекулы БМ содержат специфические центры связывания с другими молекулами БМ и с клеточными мембранами. Это обеспечивает высоко упорядоченное позиционирование молекул в БМ. Интегрины служат не только для механической связи клетки с БМ, но также и для передачи регуляторных сигналов, причем в двух направлениях - из БМ в клетку и из клетки в БМ.
Фиброретикулярный межклеточный матрикс (стромальный матрикс), с которым контактируют БМ (за исключением БМ почечных клубочков), в общих чертах сходен с БМ, но отличается от них по набору молекул и имеет менее упорядченную структуру. В частности, в отличие от БМ, в стромальном матриксе преобладают фибриллообразующие коллагены - в основном коллаген I, а также коллагены II, III, V и XI. Ламинины, характерные для БМ, отсутствуют в стромальном матриксе; вместо них здесь содержатся фибронектины.
В геноме человека один ген пептидной цепи фибронектина, но в результате альтернативного сплайсинга, а также постранляционной модификации (гликозилирование, фосфорилирование, сульфирование) образуется несколько форм белка. Фибронектин представляет собой димер двух одинаковых или немного различающихся субъединиц, соединенных антипараллельно двумя дисульфидными связями в области С-концов. Пептидная цепь образует несколько глобулярных доменов. Молекула фибронектина содержит специфические центры связывания с другими молекулами фибронектина, с коллагеном, гепарансульфатами, интегринами. Фибронектины синтезируются и секретируются многими клетками, включая фибробласты, гладкомышечные, эндотелиальные и эпителиальные клетки.
Нарушение высокоупорядоченной многомолекулярной структуры БМ (например, вследствие гликирования белков) приводит к изменению связей между молекулами БМ и внеклеточным доменом интегринов. Сигнал о повреждении с помощью интегринов передается в клетки, которые реагируют изменением ряда функциональных свойств, в том числе начинают синтезировать трансформирующий фактор роста - цитокин, стимулирующий синтез и подавляющий деградацию компонетов межклеточного матрикса.
Трансформирующий фактор роста (ТФР) - белок с молекулярной массой 12,5 кДа, гомодимер с дисульфидными связями между пептидными цепями. Он синтезируется большинством, если не всеми, клетками организма. Рецепторы ТФР - это трансмембранные Сер/Тре киназы. ТФР по аутокринному и паракринному механизмам активирует синтез компонентов матрикса - коллагенов, фибронектина, ламинина, протеогликанов, а также пролиферацию и дифференцировку клеток многих типов. С другой стороны, он снижает синтез протеаз и увеличивает содержание ингибиторов протеаз, в результате подавляется деградация компонентов межклеточного матрикса. Кроме того, ТФР стимулирует экспрессию интегринов, и тем самым способствует образованию макроструктур БМ. Таким путем обеспечивается рост БМ и фиброретикулярного межклеточного матрикса, необходимый при пролиферации клеток в норме, а также при репарации повреждений тканей.
Примером репарации повреждений может служить заживление кожной раны. В месте повреждения происходит агрегация тромбоцитов, из гранул которых освобождается ТФР-, а также тромбоцитарный фактор роста. Оба эти цитокина по паракринному механизму индуцируют секрецию ТФР- клетками в области повреждения, и таким путем инициируется процесс репарации повреждения. ТФР- служит аттрактантом для фибробластов и индуцирует синтез коллагенов и других компонентов матрикса в привлеченных фибробластах и в клетках, контактирующих с БМ. Во взаимодействии с ТФР- в репарации повреждения участвуют и другие цитокины. Тромбоцитарный фактор роста, а также фактор некроза опухолей и интерлейкин-1 индуцируют пролиферацию и миграцию клеток по мере образования матрикса. Фактор роста фибробластов индуцирует образование новых сосудов.
Как уже отмечено, тромбоцитарный фактор роста стимулирует экспрессию ТФР-. С другой стороны, ТФР- блокирует действие тромбоцитарного фактора, подавляя экспрессию его рецепторов. Возможно, такое взаимодействие имеет значение для замедления и выключения процесса на завершающих стадиях репарации.
При сахарном диабете в условиях непрерывного действия патогенного фактора (высокая концентрация глюкозы и гликирование белков) происходит дефектное ремоделирование БМ, главным образом, вероятно, вследствие постоянной секреции ТФР-: нарушается баланс между синтезом и распадом компонентов базальной мембраны в сторону усиления синтеза, нарушаются нормальные пропорции в содержании компонентов БМ и его структурная организация. Утолщение базальных мембран капилляров - один из самых ранних и постоянных признаков сахарного диабета. В области фиброретикулярного межклеточного матрикса диабетические повреждения тоже приводят к накоплению компонентов матрикса в результате активации фибробластов, синтезирующих коллагены и другие компоненты матрикса. При этом клетки пораженного органа замещаются рубцовой соединительной тканью (другие термины для описания такого процесса - фиброз, склероз).
Разные органы имеют специфические особенности молекулярного состава и структуры межклеточного матрикса, и, понятно, разный клеточный состав и функции. Поэтому диабетические повреждения матрикса, одинаковые в своей молекулярной основе в начальных стадиях, развиваются характерными для каждого органа путями.
Диабетические макроангиопатии. Поражения крупных и средних сосудов сердца, мозга, нижних конечностей обычно имеют форму атеросклероза, однако развиваются в гораздо более раннем возрасте, чем у лиц, не страдающих диабетом. Смертность от сердечно-сосудистых заболеваний при диабете примерно втрое больше, чем при других формах сердечно-сосудистых заболеваний.
Большинство патологических изменений артерий происходит в интиме. Повреждение в результате гликирования может начинаться с БМ интимы: цитокиновые сигналы приводят к изменению реактивности клеток (эндотелиальных, гладкомышечных, макрофагов), начинается поглощение липопротеинов и образование бляшки. Этому способствует хронически повышенное содержание ЛОНП (атерогенные ЛП) в крови больных диабетом.
Возможен и другой механизм повреждения артериальной стенки при сахарном диабете - гликирование белков, в частности коллагена и эластина, в среднем (media) и наружном (adventitia) слоях. Механические свойства упорядоченных сетевых структур, построенных из коллагена и эластина, имеют решающее значение для функционирования артерий и точно подогнаны к гемодинамическим условиям в кровотоке каждого участка артериального русла.
Коллаген и эластин - очень медленно обменивающиеся белки, и поэтому велика вероятность накопления в них повреждений, связанных с гликозилированием. После инкубации в течение нескольких дней отрезков артерий в растворе глюкозы в них обнаруживаются ППГ-белки, в том числе коллаген и эластин, снижается прочность и растяжимость артериальной стенки. Недавно обнаружен ППГ, обозначенный как NFC-1 (его строение пока неизвестно). NFC-1 с высокой активностью образует поперечные сшивки между молекулами коллагена. В аорте больных сахарным диабетом количество поперечных сшивок, образованных NFC-1, увеличивается с возрастом, и достигает величин до одной сшивки на одну молекулу коллагена. Это, конечно, может существенно изменить физические свойства сосуда. Однако не исключаются и нарушения, связанные с изменением скорости синтеза и деградации компонентов матрикса. Например, относительное количество гепарансульфата в средней оболочке коронарных артерий у больных сахарным диабетом снижено почти вдвое по сравнению с нормой.
Микроангиопатии. Нефропатия - одна из основных форм диабетических микроангиопатий.
Нефропатия бывает примерно у трети больных ИЗСД. Основной характеристикой диабетической нефропатии на завершающих стадиях является гломерулосклероз и нефросклероз, приводящие к хронической почечной недостаточности и к гибели больных от уремии. Клинические признаки нефропатии появляются через 10 - 15 лет после манифестации диабета, и еще в течение нескольких лет болезнь развивается до финальных состояний с появлением симптомов уремии. Диабетическая нефропатия - одна из главных причин инвалидизации и смертности больных сахарным диабетом.
Основу капиллярной стенки в клубочках составляет базальная мембрана клубочков (БМК). На внутренней поверхности БМК располагаются эндотелиальные клетки, на внешней - подоциты. Фильтрация плазмы происходит через фенестры (окна) – промежутки между эндотелиальными клетками на внутренней поверхности капилляра, и между отростками подоцитов – на наружной поверхности. Между капиллярами находится мезангий, имеющий древовидную форму и поддерживающий капилляры. Мезангий содержит мезангиальные клетки и мезангиальный матрикс. Мезангиальные и эпителиальные клетки синтезируют и секретируют компоненты мезангиального матрикса и БМК.
Основная функция почечного клубочка - обеспечить достаточную скорость фильтрации плазмы и в то же время жестко ограничить прохождение альбумина и других белков плазмы. И то, и другое определяется свойствами БМК. Плотность укладки молекул коллагена IV типа и протеогликанов определяет избирательность фильтрации по размеру фильтрующихся молекул. Гепарансульфатные цепи ГСПГ содержат много сульфатных групп, ионизированных при физиологических значениях рН. Отрицательный заряд этих молекул при фильтрации плазмы крови обеспечивает избирательную проницаемость БМК для белков в зависимости от их заряда. Альбумин человека, имеющий молекулярную массу 66 кДа (эллипсоид размером 38х150 ангстрем) и отрицательный заряд (-18 при рН 7,4), в норме лишь в небольших количествах пересекает БМК и попадает в первичную мочу. Профильтровавшийся альбумин затем эндоцитируется тубулярными клетками. Т о альбуминурия является следствием главным образом нарушения проницаемости БМК, но определенный вклад может вносить и нарушение функции тубулярных клеток.
Базальные мембраны почечных клубочков - очень интенсивно функционирующая структура: у человека за сутки фильтруется через них 180 л плазмы крови, т. е. вся жидкость организма четырежды за сутки проходит через БМК, и БМК выступает главным функциональным элементом клубочка. Канальцы нефрона тоже интенсивно функционирующие структуры, только поток воды и растворенных в ней веществ идет в обратном направлении - из первичной мочи в кровь, причем ряд веществ реабсорбируется против градиента концентрации. Однако масса канальцевой зоны значительно больше массы клубочков. Т о БМК в большей мере подвержена риску повреждения, чем другие органы, поскольку вместе с плазмой через нее идет массивный поток токсичных веществ, включая ППГ. Вероятно, гомеостаз БМК в норме поддерживается соответствующей скоростью репарации повреждений. К сожалению, нет достаточных сведений о скорости обновления белков БМК.
Считают, что при диабете повышенная в течение многих лет концентрация ППГ приводит к утолщению стенки кровеносных сосудов, экспансии мезангиального матрикса, утолщению базальных мембран. По результатам исследований гломерулярных клеток в культуре и клубочков от крыс со стрептозотоциновым диабетом можно заключить, что при диабете индуцируется экпрессия мРНК коллагенов I. III, IV и VI типов, фибронектина, ламинина, и снижается синтез мРНК гепарансульфатных протеогликанов. Уменьшение содержания гепарансульфатов относительно других компонентов ведет к нарушению структурной организации БМК и увеличению ее проницаемости для белков.
При инкубации мезангиальных клеток крысы с экзогенными ППГ активируется экспрессия мРНК коллагена IV, ламининов, гепарансульфата. Если здоровым мышам ввести ППГ-альбумин (мышиный), то увеличивается содержание компонетов межклеточного матрикса и активируется экспрессия мРНК ТФР-. Мышам линии с диабетом и с повышенным содержанием гликированного альбумина в крови вводили мышиные моноклональные антитела, специфичные к мышиному гликированному альбумину. Через 4 недели наблюдали заметное снижение протеинурии, экспансии мезангиального матрикса и экспрессии мРНК коллагена IV и фибронектина. Т о циркулирующий ППГ-альбумин хронически повреждает клубочки, которые отвечают гиперпродукцией матрикса, опосредованной ТФР-.
Накопление белков межклеточного матрикса и изменение их состава в гломерулярной, тубулярной и итерстициальной областях почки, утолщение базальной мембраны клубочков, гипертрофия и, в меньшей мере, ускоренная пролиферация мезангиальных клеток - основные патоморфологические изменения при диабетической нефропатии. При усиленном образовании межклеточного матрикса происходит прогрессивное утолщение стенки сосудов, снижение скорости клубочковой фильтрации, нарушение проницаемости базальной мембраны (и как следствие - альбуминурия). В конечном счете происходит полное закрытие сосудов и образование рубца на месте клубочка (гломерулосклероз). Сходные изменения происходят и в тубулярной области (тубулоинтерстициальный фиброз). Эти процессы характеризуют финальные стадии развития нефропатии.
Указанные изменения рассматриваются как результат нарушения репаративных процессов, направленных на устранение повреждений БМК и мезанг матрикса, вызванных гипергликемией и др факторами, действующими при сахарном диабете. Важнейшим звеном нарушения является гиперпродукция ТФР-.
У крыс со стрептозотоциновым диабетом на 24 - 40 неделях ТФР- обнаруживается в мезангии и стенках клубочковых капилляров (иммуногистохимическим методом), причем его нарастание коррелирует с альбуминурией и с накоплением коллагена I типа. Инкубация мезангиальных клеток или клубочков in vitro в среде с высокой концентрацией глюкозы резко увеличивает в них синтез ТФР-. Причинами накопления ТФР-b могут быть увеличенная секреция резидентными клетками или дегрануляция тромбоцитов. В ряде работ отмечено увеличение содержания мРНК и белка ТФР- в клубочках при экспериментальном диабете, а также при диабете у человека. Экспрессия мРНК ТФР- в клубочках диабетических крыс через 12 - 15 недель повышается в 2 - 3 раза по сравнению с контрольными крысами. В этих условиях лечение крыс инсулином снижает экспрессию мРНК ТФР-.
При остром гломерулонефрите увеличение экспрессии ТФР- и продукции матрикса бывают преходящими. Прогрессирующее накопление матрикса и развитие фиброза требует повышенной секреции ТФР- в течение длительного времени, что имеет место при диабетической нефропатии у человека и наблюдается в экспериментальных моделях диабетической нефропатии.
ДН развивается только у трети больных сахарным диабетом. Это указывает на то, что кроме гиперглюкоземии имеют значение и другие факторы, связанные с индивидуальными генетическими особенностями.
Ретинопатия обнаруживается у 30-90% больных диабетом и часто ассоциирована с нефропатией. В возрастной группе 20 - 70 лет сахарный диабет занимает первое место среди причин слепоты. При этом на долю диабетической ретинопатии приходится 70% случаев, далее следуют катаракта и другие диабетические повреждения глаза. Диабетическая ретинопатия проявляется расширением вен сетчатки, аневризмами, отеком, затем происходит новообразование сосудов в сетчатке, стекловидном теле, нарушения молекулярной структуры хрусталика. Причиной слепоты являются кровоизлияния из вновь образованных сосудов в сетчатку или в стекловидное тело и отслойка сетчатки.
Литература
1. Алексеев Л.П., Дедов И.И., Зилов А.В., Болдырева М.Н., Демидова И.Ю., Трофимов Д.Ю., Хаитов Р.М. 1998. Межпопуляционный подход в установлении ассоциированной с HLA генетической предрасположенности к инсулинзависимому сахарному диабету. Сахарный диабет 1:19-21.
2. Дедов И.И. 1998. Сахарный диабет в Российской Федерации: проблемы и пути решения. Сахарный диабет 1:7-18
3. Вахрушева Л.Л., Смирнов В.В., Галыбин А.А. и др. Иммунологические аспекты сахарного диабета у детей и в эксперименте. //Актуальные вопросы детской эндокринологии: Сб.науч.трудов. - Томск., 1990. - с. 17-18.
4. Гриневич Ю.А., Чеботарев В.Ф. Иммунобиология гормонов тимуса. - Киев., 1989. - с.103-125.
5. Гуткина О.Н. Иммуномодулирующие и метаболические эффекты тактивина при сахарном диабета: Автореф. дисс. канд. мед. наук. - Нижний Новгород., 1993. - 22 с.
6. Дорошенко Е.О. Сывороточная тимическая активность и её коррекция в комплексном лечении сахарного диабета у детей: Автореф. дисс. канд. мед. наук. - Москва., 1995. - 25 с.
7. Асташкина С.А., Калитис И.А., Гус М.И., Адоян Б.Н. Изучение остаточной секреции b-клеток у детей больных инсулинозависимым сахарным диабетом.//II Всесоюзная конференция педиатров-эндокринологов: Тез. докл. - М., 1988. - с. 15.
© 2009 База Рефератов