
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Дипломная работа: Химия и технология штатных бризантных взрывчатых веществ
Дипломная работа: Химия и технология штатных бризантных взрывчатых веществ
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ
Факультет естествознания, географии и туризма
Кафедра химии и хим. технологии неорганических веществ
ДИПЛОМНАЯ РАБОТА
на тему:
«Химия и технология штатных бризантных взрывчатых веществ»
Санкт-Петербург 2009
Содержание
Введение
1. Штатные бризантные взрывчатые вещества
1.1 Тротил: производство в военное время и сегодня, свойства тротила
1.2 ТЭН (тетранитрат пентаэритрита): химизм получения и области применения, свойства, технология производства
1.3 Гексоген: свойства и технология производства
1.4 Октоген: свойства, способы получения
2. Методическая разработка факультативных занятий по химии
Заключение
Литература
Приложение
Введение
Химические соединения или их смеси, которые содержат в молекулах определенный запас химической энергии, называют энергонасыщенными веществами. Эта энергия под действием внешнего импульса в результате протекания химических реакций освобождается, превращаясь в тепловую, световую, механическую, электрическую и т.д.
Наиболее распространенные типы энергонасыщенных веществ – это взрывчатые вещества, пороха и компоненты твердых ракетных топлив, пиротехнические составы. В данной дипломной работе речь пойдет о штатных бризантных взрывчатых веществах.
Преобразование химической энергии в другие виды энергии во взрывчатых веществах осуществляется в результате чрезвычайно быстро протекающей реакции химического взрыва. Энергия взрыва может быть использована в самых разных целях.
Основная особенность взрывчатых веществ, которая и обусловила появление термина «энергонасыщенные вещества» – это экзотермичность реакций взрыва, сопровождающихся выделением большого количества теплоты, которое разогревает газообразные продукты превращения этих веществ до высокой температуры (3000–5000 К). Чем больше количество теплоты выделяется в результате взрыва единицы массы вещества, тем, как правило, более эффективно действие взрывчатого вещества, т.е. количество теплоты, выделяемое при химической реакции – основной критерий работоспособности.
Взрывчатые вещества обычно делят на бризантные и инициирующие взрывчатые вещества. Бризантные взрывчатые вещества нашли широкое применение в технике и народном хозяйстве в качестве мощных и компактных источников механической энергии. Примером бризантных взрывчатых веществ являются такие производимые промышленностью соединения, как тротил, гексоген, ТЭН, октоген.
Механическая работа, являющаяся основной целью взрыва, совершается за счет той потенциальной энергии, которой обладает заряд взрывчатого вещества.
Ввиду относительно высокой стоимости взрыва важно, чтобы его энергия была использована наиболее эффективно. Говоря о работоспособности зарядов взрывчатых веществ, обычно различают бризантное (дробящее) и фугасное (общее) механическое действие взрыва. Бризантное действие проявляется в непосредственной близости от заряда взрывчатого вещества. На бризантные формы работы затрачивается незначительная часть энергии взрыва.
К недостаткам взрывчатых веществ следует отнести недостаточно высокие взрывчатые характеристики тротила и довольно высокую опасность в обращении с остальными тремя штатными взрывчатыми веществами, а также плохую прессуемость последних.
Взрывчатые вещества как высококонцентрированный и экономичный источник энергии широко применяют в различных отраслях народного хозяйства. Около 90% всего объема руд цветных и черных металлов в нашей стране добывают взрывным способом. Массовые взрывы широко используются при вскрытии рудных тел, угольных пластов и месторождений других полезных ископаемых, в строительстве, при сооружении плотин и насыпей, прокладке авто- и железнодорожных магистралей, водных каналов, спрямлении русел рек, прокладке нефте- и газопроводов, особенно в труднодоступной для техники местности, при проведении тоннелей, прокладке шахтных стволов и других горных выработок.
Взрывчатые вещества также широко применяют при взрывных способах обработки в машиностроении и металлургии – штамповке, сварке, изготовлении биметаллических листов, упрочнении деталей машин, резании металлов; при перфорации нефтяных скважин, при тушении лесных пожаров, уплотнении грунтов, в гидромелиоративном строительстве, расчистке и выравнивании местности и для других технических нужд.
Продолжается поиск и исследование дальнейших путей использования и управления энергией взрыва. В настоящее время применяется взрывной способ производства некоторых особенно ценных минералов и искусственных материалов, ускоряются отдельные химические процессы с использованием сверхвысоких давлений взрыва, проводятся работы по искусственному дождеванию, внедряются методы взрывного бурения.
Целью данной работы является: на основе доступных литературных источников произвести обзор свойств и технологий получения штатных бризантных взрывчатых веществ; разработать факультативное занятие по теме «Бризантные взрывчатые вещества» для учащихся старших классов средней общеобразовательной школы.
1. Штатные бризантные взрывчатые вещества
1.1 Тротил: производство в военное время и сегодня, свойства тротила
Тротил (тринитротолуол) широко используется самостоятельно и как компонент взрывчатых составов в военном деле для снаряжения боеприпасов, в мирных целях как промышленное взрывчатое вещество. Преимущество тротила перед другими индивидуальными ВВ обусловлено благоприятным сочетанием физико-химических, взрывчатых и технологических свойств.
Первые промышленные установки производства тротила в России были созданы в 1909 году на Охтинском, в 1912 году на Самарском (ныне ОАО «Полимер»), в 1922 году на Нижегородском (ныне ГУП «Завод им. Я.М. Свердлова») заводах по технологии фирмы «Карбомит» (Германия). В дальнейшем производство тротила развивалось на базе отечественных разработок и характеризовалось весьма высокими темпами, что позволило увеличить объем его выпуска в послевоенные годы в 11 раз [5, 113].
Широкое применение тротила в промышленности является характерной особенностью России. При этом доля тротила, используемого для снаряжения боеприпасов, в общем объеме производства в послевоенные годы не превышала 10%.
Первые промышленные установки производства тротила в России были весьма несовершенны. Получение тротила осуществлялось нитрованием толуола в три стадии с последующей очисткой тротила-сырца перекристаллизацией из этилового спирта. Периодический способ на всех технологических операциях, отсутствие кислотооборота на стадии нитрования, ручной межфазный транспорт приводили к повышенным трудовым и материальным затратам. В 1932–1933 годах был разработан и внедрен горячий кислотооборот, изменен порядок дозировки компонентов – применена дозировка нитруемого соединения к нитрующей смеси. Несмотря на значительное улучшение технико-экономических показателей периодический процесс не удовлетворял требованиям бурно развивающейся
индустриализации страны, в том числе требованиям промышленности боеприпасов. Поэтому уже в 1936 году на Чапаевском химзаводе был освоен непрерывный четырехфазный противоточно-прямоточный способ нитрации толуола до тротила-сырца с горячим кислотооборотом.
В 1940 году в промышленном масштабе на заводе им. Я.М. Свердлова был освоен другой вариант непрерывного процесса – многофазный противоточный способ нитрации, созданный заводскими инженерами А.Т. Васильевым, Н.П. Кошелевым, Г.М. Васильевым.
Переход на сульфитную очистку вместо кристаллизации из этилового спирта был осуществлен в 1933–1937 годах по предложению А.Т. Васильева, П.И. Канавца, И.А. Мазеля. Главные преимущества использования сульфита натрия – это повышение безопасности за счет исключения применения легковоспламеняющегося растворителя – этилового спирта, увеличение выхода очищенного тротила, то есть снижение расхода сырья и улучшение технико-экономических показателей. Процесс очистки тротила сульфитом натрия длительное время оставался периодическим, хотя к этому времени был осуществлен перевод операций сушки и чешуирования на непрерывный режим.
Новый этап в развитии технологии и производства тротила начался в послевоенный период (1945–1960), когда значительно увеличилась потребность в промышленных взрывчатых веществах для добывающих отраслей промышленности. Наиболее значительными из результатов исследований, решивших выбор технологии в пользу противоточного метода, явились две разработки – безолеумный процесс (П.И. Канавец, Т.Н. Серебрянникова, В.В. Гисин, НИИ-6; А.Т. Васильев, Г.М. Васильев, завод им. Я.М. Свердлова), освоенный в 1948 году и изобретение в 1947 году конического шнек-подъемника и нитраторов с совмещенными или выносными сепараторами, расположенными выше нитраторов (А.И. Борисов, В.М. Елецкий).
В дальнейшем совершенствование технологических линий с одновременным повышением их производительности шло в направлении использования более крепких кислотных смесей вплоть до 100%-ной серной кислоты, а также за счет распределения дозировки серного компонента в конец и в середину системы. Так завершился определенный этап создания высокопроизводительных безопасных технологических линий нитрования толуола до тротила, превосходящих для своего времени по техническому уровню известные западные технологии – периодические в Германии, США, непрерывные, но с низкой производительностью и сложным аппаратурным оформлением в Англии, Испании, Италии.
Производство тротила
Процесс получения тротила складывается из следующих стадий:
1) Нитрование толуола до тротила;
2) водная промывка тротила от кислоты;
3) чистка тротила от примесей;
4) сушка тротила.
Нитрование толуола до тротила
Изучение реакции нитрования толуола до мононитротолуола было направлено главным образом на снижение выхода мета-нитротолуола, чтобы в последующем получить тротил с меньшим содержанием примесей. Были исследованы также и некоторые характеристики нитрования толуола в гетерогенных условиях: растворимость, распределение компонентов между слоями, влияние перемешивания на скорость реакции и т.д.
Коэффициент распределения азотной кислоты между толуольным и сернокислотным слоями (при 5°С и концентрации H2S04 70%) равен 0,066, при более низкой концентрации H2S04 он равен нулю. Это указывает на то, что азотная кислота при гетерогенном нитровании толуола лишь в незначительной степени переходит в органический слой и поэтому доля протекающей там реакции практически равна нулю.
Низкая растворимость толуола в серной кислоте умеренных концентраций, отсутствие перехода азотной кислоты в органический слой, а также резкая зависимость скорости реакции нитрования толуола от интенсивности перемешивания и объемной доли минерального слоя позволяют предположить, что реакция нитрования толуола в гетерогенных условиях протекает возле поверхности раздела слоев. Скорость в этом случае будет определяться концентрацией реагирующих компонентов на этой поверхности, которая в свою очередь определяется скоростью диффузии реагирующих компонентов из глубины слоя к поверхности раздела и скоростью отхода от нее продуктов реакции.
Указанные процессы, а также состояние реагирующих компонентов, зависят от температуры, концентрации кислотной смеси и интенсивности перемешивания.
Производство тротила усложняется в первой ступени нитрования нежелательным образованием мета-нитротолуола. Образование 5–6% этого изомера в дальнейшем приводит к образованию 5–6% несимметричных тринитротолуолов, загрязняющих тротил.
Выход мета-нитротолуола может быть уменьшен:
1) При снижении температуры нитрования;
2) при введении в кислотную смесь нитрата натрия
Установлены следующие закономерности процесса нитрования толуола до мононитротолуола в гетерогенных условиях:
а) скорость процесса, по-видимому, определяется скоростью диффузии компонентов к зоне реакции, так как нитрование идет главным образом на поверхности раздела слоев [4, с. 212];
б) скорость нитрования в гетерогенных условиях сравнительно мало зависит от температуры, в то же время с понижением температуры снижается выход мета-нитротолуола. Следовательно, целесообразно проведение нитрования толуола при низкой температуре. Это будет способствовать снижению выхода мета-нитротолуола и сравнительно мало повлияет на скорость нитрования;
в) целесообразно применение наиболее интенсивного перемешивания с целью увеличения скорости реакции, особенно при низкотемпературном режиме нитрования. Это приведет к увеличению производительности системы.
Нитрование мононитротолуола до динитротолола
Исследованием реакции нитрования мононитротолуола до динитротолуола занимались мало, что до некоторой степени понятно, так как в производстве тротила эта стадия является средним звеном между первой и третьей. Из первой стадии туда поступает мононитротолуол, а из третьей серная кислота в виде отработанной кислоты. Нитрование мононитротолуола до динитротолуола обычно проводят, используя всю отработанную кислоту от третьей стадии и крепкую или слабую азотную кислоту. Больших затруднений в производстве эта стадия не составляет.
Скорость реакции нитрования мононитротолуола в гетерогенных условиях, также как и толуола, зависит от интенсивности перемешивания (величины поверхности раздела слоев). Однако эта зависимость менее резкая, чем для толуола. Зависимость скорости нитрования мононитротолуола в гетерогенных условиях от интенсивности перемешивания указывает на то, что нитрование в значительной степени протекает возле поверхности раздела слоев. Наряду с этим идет нитрование и в минеральном слое, где концентрация мононитротолуола в условиях процесса, достаточно высокая.
Нитрование динитротолуола до тринитротолуола
Нитрование динитротолуола является наиболее медленной стадией процесса получения тротила вследствие резкого торможения скорости вступления третьей нитрогруппы двумя другими нитрогруппами, уже имеющимися в бензольном ядре. Это наглядно видно на примере нитрования динитротолуола концентрированной азотной кислотой, которая взаимодействует с ним с незначительной скоростью. Повышение температуры мало изменяет эту скорость, а лишь способствует развитию сильных окислительных процессов. Серно-азотные смеси, особенно концентрированные, нитруют динитротолуол с большей скоростью, чем чистая азотная кислота.
В производстве тротила нитрование динитротолуола, так же как толуола и мононитротолуола, осуществляется в гетерогенных условиях. Скорость процесса в этом случае складывается из скоростей процессов диффузии реагирующих компонентов из одного слоя в другой и затем нитрования. Общая скорость определяется скоростью наиболее медленного процесса. Если скорость нитрования больше скорости диффузии, то обычно реакция протекает на поверхности раздела, как это имеет место для случая нитрования толуола и мононитротолуола. При малой скорости нитрования реакция будет происходить в объеме того слоя, в котором имеются реагирующие компоненты.
Динитротолуол хорошо растворяется в серной кислоте, тротил растворяется в ней значительно хуже.
Растворимость сплавов динитротолуол-тринитротолуол лежит между величинами растворимости чистого динитротолуола и чистого тринитротолуола и зависит от состава сплава. С увеличением содержания тротила растворимость сплава уменьшается. Значительное снижение растворимости наблюдается при добавлении к динитротолуолу 20–30% тротила. Дальнейшее изменение растворимости происходит более плавно и снижается пропорционально увеличению содержания тротила в сплаве.
Растворимость продуктов сильно зависит от концентрации серной кислоты и значительно меньше от температуры.
При частичном растворении сплава динитротолуол-тротил происходит распределение динитротолуола между слоями. Динитротолуол как продукт, обладающий большей растворимостью, в большем количестве переходит в минеральный слой, чем тротил. Таким образом, минеральный слой по сравнению с органическим обогащается динитротолуолом. Тем не менее, коэффициент распределения динитротолуола вследствие низкой растворимости сплавов в серной кислоте очень мал (0,3–0,4), что указывает на сравнительно небольшую концентрацию динитротолуола в минеральном слое.
Коэффициент распределения азотной кислоты между минеральным и органическим слоями для случая нитрования динитротолуола в среде 93% H2SO4 при 90°С равен примерно единице, что указывает на большую степень поглощения азотной кислоты органическим слоем [18, с. 106].
В гомогенных и гетерогенных условиях с увеличением концентрации HNO3 в кислотной смеси скорость реакции увеличивается до предела, а затем падает. В гетерогенных условиях максимум отодвигается в сторону смесей, содержащих больше HNO3. Подсчет концентрации HNO3 в минеральном слое по коэффициенту распределения показывает, что она равна той же величине, что и концентрация HNО3, при которой скорость нитрования в гомогенной среде достигает максимума.
Таким образом, при нитровании в гетерогенных условиях снижение концентрации HNО3 в минеральном слое уменьшает скорость нитрования динитротолуола, основная масса которого нитруется, по-видимому, в минеральном слое.
Нитрование толуола до мононитротолуола и мононитротолуола до динитротолуола в гетерогенных условиях в значительной степени является «поверхностной» реакцией, что следует из зависимости скорости ее от интенсивности перемешивания. Реакция нитрования динитротолуола в гетерогенных условиях не ограничивается одной поверхностью раздела (о чем свидетельствует малая зависимость ее скорости от интенсивности перемешивания), а значительно распространяется вглубь минерального слоя (основная часть реакции, протекает в минеральном слое). Реагирующие компоненты – динитротолуол и азотная кислота распределяются между слоями в соответствии с растворимостью в них и с соотношением объемов слоев. В случае нитрования динитротолуола реакция идет в объеме каждого слоя. Скорость нитрования в органическом слое, по-видимому, имеющая место только при высоком факторе нитрующей активности, значительно ниже скорости нитрования в минеральном слое.
Причиной этого является то, что в органическом слое находится только HNО3, так как H2SО4 в этот слой практически не переходит. Поэтому при сравнительно малых объемах органического слоя можно считать, что нитрование динитротолуола идет только в минеральном слое, причем скорость его зависит от степени растворимости динитротолуола в кислоте. С повышением концентрации кислоты увеличивается растворимость в ней динитротолуола и значительно облегчается процесс нитрования динитротолуола.
Присутствие органического слоя в некотором отношении отрицательно влияет на течение процесса нитрования. Органический слой обладает высокой растворяющей способностью по отношению к нитрующему агенту – азотной кислоте, что в значительной степени снижает концентрацию азотной кислоты в минеральном слое, снижая тем самым скорость нитрования.
Вторичная реакция – окисление протекает в органическом и минеральном слоях. При этом, по-видимому, окисление в органическом слое вследствие присутствия в нем HNО3 без H2S04 идет в большей степени, чем в минеральном. Окислительное действие азотной кислоты в минеральном слое снижается присутствием серной кислоты.
Отрицательное влияние на скорость нитрования органического слоя сказывается также и на снижении концентрации динитротолуола в минеральном слое. Снижение концентрации динитротолуола происходит за счет перехода его в органический слой, правда только в том случае, если последний представляет собой расплавленный тротил. В присутствии слоя расплавленного тротила динитротолуол распределяется между органическим и минеральным слоями в соответствии с растворимостью в них и соотношением объемов слоев. По мере течения реакции и увеличения слоя тротила уменьшается количество динитротолуола, растворенного в нитросмеси.
Отрицательное влияние тротила на скорость нитрования несколько смягчается в конце процесса, когда в сплаве остается небольшое количество недонитрованного динитротолуола. В этих условиях, очевидно, относительная доля динитротолуола в минеральном слое больше вследствие того, что этот слой по отношению к динитротолуолу благодаря малому количеству его становится ненасыщенным [11, с. 159].
Таким образом, наиболее благоприятны условия гетерогенного нитрования динитротолуола в начале процесса, когда органический слой состоит из чистого динитротолуола. В этих условиях скорость процесса будет максимальной.
Наименее благоприятны условия нитрования в конце процесса, когда действующая масса динитротолуола в минеральном слое мала. Чтобы увеличить скорость нитрования в конце процесса, необходимо его вести при более высокой температуре либо применять концентрированную кислотную смесь. В начале процесса вследствие значительной концентрации динитротолуола возможно применение слабых кислотных смесей и осуществление нитрования при более низкой температуре. Таким образом, в этом процессе целесообразен противоток между нитруемым соединением и нитрующей кислотной смесью.
Реакция нитрования динитротолуола в гетерогенных условиях протекает в основном в минеральном слое, следовательно, скорость ее определяется объемом этого слоя и концентрацией в нем реагирующих компонентов. Последние могут быть определены, исходя из коэффициента распределения этих компонентов между минеральным и органическим слоями.
Промывка тротила от кислоты
Тротил, полученный в мастерской нитрации, содержит 3–5% кислоты, от которой его необходимо освободить. Кислоту, примешанную к тротилу вследствие неполной сепарации, а также и растворенную в нем, удаляют промыванием водой. Применение щелочи для нейтрализации кислоты при окончательной промывке не допускается вследствие возможности получения высокочувствительных и малостойких металлических производных тротила.
Тротил промывают горячей водой обычно в расплавленном состоянии, при этом, кроме минеральной кислоты, в раствор переходит также часть продуктов побочных реакций, например, тринитрокрезол, тринитробензойная кислота. Последняя при горячей промывке частично переходит в тринитробензол [18, с. 118].
В Германии промывку тротила производили путем смешивания расплавленного продукта с промывной жидкостью и последующей декантацией в том же аппарате. Промывные аппараты имеют ту же конструкцию, что и нитраторы. Отстоявшаяся промывная жидкость эвакуируется с помощью сжатого воздуха. Тротил промывают три раза: водой, 3–4%-ным раствором соды и затем снова водой.
В США и Англии тротил промывают в чанах с мешалками и только горячей водой четыре-шесть раз до остаточной кислотности 0,01%. Потеря тротила с промывной водой обычно достигает 0,5%.
Очистка тротила
Промытый тротил (сырец), имеющий температуру затвердевания не ниже 77,4°С, содержит до 6% примесей, представляющих собой главным образом несимметричные изомеры тринитротолуола и динитротолуол, а также тетранитрометан, продукты окисления и др. Примеси вызываю понижение температуры затвердевания тротила. Поэтому тротил-сырец в дальнейшем очищают от этих примесей либо химическим способом – перевод примесей в растворимые в воде соединения, либо физическим способом – кристаллизация из растворителей или промывка кристаллического тротила растворителями.
Химический способ очистки. Этот способ очистки тротила-сырца основывается на превращении примесей путем взаимодействия их с реагентом в растворимые в воде соединения. Подобрать подобные реагенты не трудно, так как основными примесями являются несимметричные тринитротолуолы, легко замещающие нитрогруппу, стоящую в метаположении по отношению к метильной группе. Такими реагентами, могут быть, например, щелочь, аммиак, метиламин и другие аминопроизводные, реагирующие с несимметричными тринитротолуолами.
Более удобным реагентом оказался сульфит натрия, и в течение последних тридцати лет его широко используют для очистки тротила-сырца [21].
Сульфитная очистка основана на том, что сульфит натрия, реагируя с большинством примесей, полученных в результате побочных реакций и нитрования примесей, находящихся в толуоле, образует соединения, растворимые в воде и в водном растворе сульфита натрия, благодаря чему примеси легко отмываются при дальнейших операциях.
Действие водных растворов сульфита натрия на примеси различно.
С несимметричными изомерами тринитротолуола сульфит натрия реагирует на холоду, образуя динитротолуолсульфонаты натрия, хорошо растворимые в воде.
Тетранитрометан сравнительно легко взаимодействует с сульфитом натрия. Конечным продуктом является тринитрометан–вещество, хорошо растворимое в воде с желтым окрашиванием:
С (НО2)4 +Na2SО3 + Н2О→С (NО2)3 H + NaNО2 + NaHSО4.
С тринитробензолом образуется аддитивное соединение ярко-красного цвета, легко растворимое в разбавленных растворах сульфита натрия.
Продукты окисления фенольного типа также легко растворяются в разбавленных растворах сульфита, образуя феноляты.
На недонитрованные примеси тротила: динитротолуол, динитробензол, а также на тринитрометаксилол разбавленные растворы сульфита натрия не действуют. Поэтому тротил, направляемый на сульфитную очистку, должен содержать по возможности минимальное количество этих соединений.
Разбавленные растворы сульфита натрия при низких температурах на α-изомер практически не действуют, но в более концентрированных образуются аддитивные и другие соединения α-тринитротолуола с сульфитом натрия, окрашивающие раствор в ярко-красный цвет. Часть этих соединений разлагается водой с регенерацией α-тринитротолуола. Растворимость α-тринитротолуола в растворах сульфита натрия зависит от концентрации этих растворов, что видно из следующих данных: 3%-ный раствор сульфита натрия при 20°С растворяет 0,3% α-тринитротолуола; 6%-ный раствор сульфита натрия при 20°С растворяет 0,6% α-тринитротолуола; 12%-ный раствор сульфита натрия при 20°С растворяет 2,3% α-тринитротолуола [23, с. 72].
Следовательно, уже 6%-ный раствор является довольно сильным растворителем, приводящим к большим потерям тротила.
Значительное влияние на потери тротила оказывает и температура обработки его раствором сульфита.
Так при действии 250 мл 5%-ного раствора Na2S03 на а-тринитротолуол при 30, 40, 50 и 60° общие потери составляют 0,9; 1,1; 2,0; 5,0 г, а потери нерегенерируемые – 0,4; 0,6; 1,5 и 5,0 г соответственно.
Следовательно, потери α-изомера с повышением температуры выше 40°С значительно возрастают, причем переходят в потери не регенерируемые с разбавлением раствора, что говорит о новом характере получающихся соединений. Минимальная потеря α-тринитротолуола имеет места при обработке его сульфитом в течение максимум одного часа при температуре ниже 40°С и разбавлении сразу же после конца реакции равным объемом воды. Поэтому, принимая во внимание указанное выше влияние концентрации сульфитного раствора и температуры обработки, на заводах работают с разбавленными растворами – от 2 до 5% активного сульфита, проводя обработку преимущественно при температуре ниже 60°С. При применении более разбавленных растворов сульфита натрия, концентрации около 2%, возможно проведение очистки и при 75°С; потери α-изомера при этом сравнительно небольшие [24, с. 31].
Обработка сульфитным раствором дает лучшие результаты, если ей подвергается тротил в виде кристаллов или в крайнем случае в виде раздробленных мелких гранул. Так как при образовании кристаллов жидкие примеси собираются в виде тонкой пленки на их поверхности, то они легко подвергаются действию сульфита, в то время как в расплавленном тротиле или в крупных гранулах примеси распределены по всей массе и таким образом как бы предохранены от воздействия сульфита. По указанной причине в США и Германии промывке тротила сульфитом натрия предшествует кристаллизация расплавленного тротила под водой.
Основными аппаратами мастерской сульфитной очистки являются кристаллизаторы и воронки.
Воронка служит для отжимания и отмывания сульфитного раствора и растворенных в воде примесей, образовавшихся вследствие обработки тротила сульфитом натрия. Горячей водой отмываются также легкоплавкие примеси к тротилу, выделившиеся на поверхность кристаллов во время кристаллизации (динитротолуол, динитробензол).
В кристаллизатор при работающей мешалке заливают горячую воду (с температурой не ниже 80°С) и расплавленный тротил (соотношение 1:1 по объему). Для охлаждения пускают в ход вентиляцию. По достижении в кристаллизаторе температуры 56–58°С приливают раствор сульфита натрия (в виде 10–15%-ного раствора). В США применяют 16%-ный раствор сульфита, содержащий 0,5% бисульфита.
Во время кристаллизации из тротила выделяются примеси. Распределяются они на поверхности кристаллов чистого α-изомера, а поэтому становятся более доступными действию сульфита. Если нарушить правильность кристаллизации резким охлаждением или неравномерным перемешиванием в различные периоды кристаллизации, то могут образоваться сростки кристаллов или гранулы. Внутри этих сростков и гранул кристаллы тротила будут недоступны для промывки раствором сульфита, что снизит качество тротила и приведет к получению некондиционного продукта. Даже если в лучшем случае гранулы образуются после воздействия сульфита на примеси, они все же могут явиться причиной выхода некондиционного продукта, так как при этом будут внутри содержать маточный раствор сульфита, не поддающийся промывке водой на воронке.
Тротил из кристаллизатора вместе с сульфитным раствором спускается на воронку. Для того чтобы выгрузка шла равномерно, мешалка кристаллизатора продолжает работать до полного опорожнения аппарата, а кран, через который спускают массу, периодически прочищают медным прутом.
На воронке отсасывают сульфитный маточный раствор в специальный приемник. Затем тротил промывают несколькими порциями воды. Температура промывной воды должна быть в пределах 60–68°С. Более горячая вода может сплавить кристаллы тротила в комки, более холодная вода не расплавит примесь динитротолуола, оставшегося в виде пленки на поверхности кристаллов. То и другое повлечет за собой выход брака, так как в первом случае внутри комков останется неотмытый маточный раствор сульфита натрия, а во втором случае тротил не будет освобожден от динитропроизводных.
Кристаллы тротила на воронке промывают до получения прозрачной промывной воды (отсутствие расплавленного динитротолуола) и удовлетворительного анализа на отсутствие сульфита натрия. Температура затвердевания тротила должна быть не ниже 80,3°С.
Тротил для анализа берут из воронки (с середины по высоте), а промывную воду из сливной линии.
Важным преимуществом сульфитной очистки тротила является простая аппаратура; большинство аппаратов легко заменяемы и недороги.
Сульфитная очистка тротила менее опасная операция (особенно в пожарном отношении), чем кристаллизация из растворителя. Однако значительное скопление тротила в аппаратах главным образом из-за периодичности их работы резко снижает это преимущество. Периодичность процесса делает практически невозможным применение автоматизации контроля и управления процессом.
Физические способы очистки. Сульфитный метод очистки тротила имеет весьма существенные недостатки – большие безвозвратные потери продукта (до 10%) и образование значительного количества очень токсичной отработанной воды. Физические способы очистки тротила, основанные на перекристаллизации его из растворителя или промывке кристаллов растворителями, уменьшают безвозвратные потери. Выделенные из растворителя примеси могут быть использованы как взрывчатые вещества.
В Германии применялась перекристаллизация тротила из спирта, из толуола и из азотной кислоты [7, с. 18].
Перекристаллизация тротила из спирта является одним из наиболее давно известных способов его очистки. Предварительно тротил должен быть промыт до кондиционной кислотности.
В Германии при кристаллизации из спирта последний брали в количестве трех частей на одну часть тротила, т.е. продукт полностью не растворялся, а осуществлялось лишь промывание его горячим спиртом и кристаллизация в присутствии спирта. Тротил отделяли от маточного раствора на вакуум-фильтре и промывали чистым спиртом, взятым в количестве 1:1.
Спирт для очистки используют 5–6 раз, а затем его передают на ректификацию. При ректификации в результате разгонки получают вновь чистый спирт и примеси тротила, которые под названием «тротиловое масло» идут для приготовления аммонитов.
При спиртовой очистке тротила имеется только два преимущества: отсутствие токсичных промывных вод и возможность использования отделенных от тротила примесей. Вместе с тем способ имеет ряд существенных недостатков. Основными являются потери дорогого растворителя, громоздкость аппаратуры, опасность возникновения пожара и взрывоопасность, а также и ряд других недостатков.
В Германии кристаллизацию тротила из толуола производили по-видимому, по той технологической схеме, что и кристаллизацию из спирта. Разница состояла в том, что тротил полностью растворяли в толуоле при 60°С. Для этого на 0,9 вес. ч. тротила использовали 1 вес. ч. толуола. Раствор охлаждали до 25°С и выкристаллизовавшийся при этом тротил отжимали на центрифуге, после чего многократно промывали. Регенерировали толуол перегонкой острым паром.
Делалась попытка осуществить очистку тротила кристаллизацией его из азотной кислоты на двух установках непрерывного действия. Очистка производилась следующим образом.
Тротил растворяли при 60°С в маточной азотной кислоте (примерно 70%-ной), взятой в количестве 3:1. Раствор тротила непрерывно подавали в кристаллизатор с мешалкой, где температура поддерживалась 25–30°С. Из кристаллизатора содержимое попадало на ленточный ячейковый вакуум-фильтр. Здесь продукт промывали последовательно крепкой 60%-ной и 30%-ной азотной кислотой, и затем теплой и холодной водой.
Промывные кислоты использовали вновь. Крепкую промывную кислоту добавляли к маточному раствору, а также применяли для растворения тротила. Избыток кислоты разбавляли водой для выделения нитропродуктов, которые шли на приготовление взрывчатых смесей. Промывную 60%-ную кислоту добавляли к 30%-ной, используемой на промывку. Промывную 30%-ную кислоту смешивали с промывными водами.
Очищенный таким образом тротил расплавляли, промывали дополнительно горячей водой, а затем высушивали.
Сушка тротила
Сушат тротил в отдельном, удаленном на необходимое с точки зрения безопасности расстояние и окруженном земляным валом, здании. Сушильные агрегаты состоят из сушильной ванны и барабана для чешуирования тротила.
Сушильные ванны представляют собой либо цилиндрические сосуды (в Германии такие же, как нитраторы), либо прямоугольные емкости, На дне этих сосудов имеются змеевик для глухого пара и воздушные барбатеры. Сушку производят продуванием через слой расплавленного, нагретого до 100°С, тротила сжатого воздуха с давлением 0,35–0,40 атм. Отработанный воздух вместе с влагой уходит в вентилятор через трубу, присоединенную к верхней части крышки. Под крышкой вмонтированы оросительные трубы дренчерной системы для тушения водой в случае воспламенения тротила.
Измеряют температуру внутри сушильных аппаратов, как правило, дистанционными термометрами. Объем сушильной ванны, работающей по указанному принципу, должен обеспечивать пребывание в ней тротила 30–40 мин. (при условии небольшой толщины слоя тротила).
В Германии сушат тротил также продувкой воздухом, но при этом аппараты держат под вакуумом (500 мм рт.ст.) и заполняют их тротилом полностью. При емкости аппарата 15 м3 продолжительность сушки доходит до 4–6 час.
Контролем сушки является температура затвердевания тротила в пробе, отбираемой на анализ. Если температура затвердевания окажется ниже требуемой (80,2°), что свидетельствует о недостаточном высыхании, уменьшают скорость прилива тротила [24, с. 48].
Высушенный тротил из ванны стекает в обогреваемое корыто под барабаном для чешуирования.
Чешуированный тротил ссыпают либо в деревянные ящики, либо в джутовые мешки.
Готовый тротил первого сорта должен удовлетворять соответствующим техническим условиям: он должен представлять однородную массу в виде чешуек желтого цвета, без посторонних видимых на глаз примесей и без признаков подмочки. Температура затвердевания его – не менее 80,2°С; содержание влаги и летучих – не более 0,07%; кислотность по H2S04 – не более 0,011%; содержание веществ, не растворимых в бензоле или толуоле, – не более 0,1% и маслянистость не более чем у эталонного образца.
Наиболее важным критерием качества тротила является температура затвердевания. Высокая температура затвердевания свидетельствует о чистоте продукта и, следовательно, его стойкости. Другим фактором, также влияющим на химическую стойкость, как самого тротила, так и оболочки снаряда, является содержание кислоты.
Определение содержания влаги и летучих веществ в тротиле характеризует взрывчатые свойства его, так как повышенное содержание влаги уменьшает восприимчивость тротила к детонации. Повышение содержания нерастворимых примесей может изменить чувствительность тротила; так, примесь песка увеличивает чувствительность к удару и трению. Маслянистость тротила в первую очередь характеризует восприимчивость его к капсюлю-детонатору [18, с. 125].
Тротил, не удовлетворяющий какому-либо из перечисленных условий, должен быть забракован. Все виды брака, не считая совершенно случайных причин (занесение ветром песка, подмочка водой и т.п.), зависят главным образом от нарушений и отклонений от правильного ведения процесса производства.
Брак по высокой кислотности бывает крайне редко. Брак по температуре затвердевания и маслянистости может получиться как из-за недостаточно высокого качества тротила, поступившего из нитрационных мастерских, так и вследствие нарушения правильности процессов обработке тротила в вакуум-воронке. Брак по маслянистости может быть также результатом плохой очистки от машинного масла воздуха, идущего на мешку в сушильную ванну.
Брак по нерастворимому остатку и цвету зависит от чистоты воды и аппаратуры. При пуске новой или некоторое время не работавшей аппаратуры цвет тротила первых партий всегда не соответствует нормальному. Брак по влажности – результат недостаточной сушки в ванне (низкая температура, плохое перемешивание, большая скорость и т.п.).
Брак по влажности может быть исправлен вторичной пересушкой, брак по температуре затвердевания, маслянистости и нерастворимому остатку – повторной кристаллизацией, а брак по цвету исправить нельзя и такой тротил идет на изготовление промышленных взрывчатых веществ.
Свойства тротила
Тротил получают нитрованием толуола. Известно шесть изомеров тринитротолуола, имеющих одну и ту же общую формулу С6Н2(NО2)3СН3, но отличающихся различным положением нитрогрупп в бензольном ядре, а вследствие этого и различными физико-химическими свойствами. Применяемый в практике тротил состоит в основном из симметричного, или α-изомера тринитротолуола.
2,4,6- или α-тринитротолуол представляет собой желтое вещество, имеющее две полиморфные кристаллические формы.
Температура затвердевания 2,4,6 – тринитротолуола – 85°С; уд. вес 1,663, расплавленного (при 82°С) 1,467. Гравиметрическая плотность кристаллического тринитротолуола 0,9–1,0 [19, с. 53].
Скрытая теплота плавления α-тринитротолуола 21,41 кал/г, теплота кристаллизации 5,6 ккал / моль, теплопроводность при 25° 0,0005 ккал/сек/см°С.
Гигроскопичность α-тринитротолуола составляет около 0,05%, поэтому при его хранении не требуется герметической укупорки.
Растворимость α-тринитротолуола в воде низкая. Так, при 15° в 100 частях воды растворяется 0,02 частей, а при 100°С в 100 частях воды растворяется 0,15 частей α-тринитротолуола.
Малая растворимость α-тринитротолуола в воде является благоприятным свойством, облегчающим водную промывку его от кислот. Тем не менее, и эта растворимость влечет за собой, с одной стороны, потери продукта и, с другой стороны, загрязнение воды. Воду с содержанием 0,15% α-тринитротолуола нельзя спускать в водоемы, поэтому до спуска в водоемы ее подвергают охлаждению и отстаиванию с целью выделения основной массы растворенного тротила.
В органиченных растворителях α-тринитротолуол растворяется достаточно хорошо, лучшими растворителями его являются: пиридин, ацетон, бензол, толуол, хлороформ. Плохо растворяется а-тринитротолуол в эфире и сероуглероде.
Основные взрывчатые свойства тротила:
Чувствительность к удару по русской пробе – 4–8% взрывов
Температура вспышки – 290°С
Расширение в бомбе Трауцля – 285 мл
Скорость детонации – 7000 м/с
1.2 Тэн: химизм получения и области применения, свойства, технология производства
Тэн (тетранитрат пентаэритрита) впервые был получен этерификацией пентаэритрита в 1894 году в Германии, однако его промышленный выпуск начался только в 1920-е годы, после того, как были разработаны рентабельные промышленные способы производства формальдегида и ацетальдегида, являющихся основным сырьем для получения пентаэритрита. Необходимость производства тэна диктовалась появлением новых средств инициирования (детонирующих шнуров и капсюлей – детонаторов), применение тэна в которых более эффективно по сравнению с другими взрывчатыми веществами и их смесями. С.П. Byколов первым изучил его взрывчатые характеристики и показал, что из эфиров азотной кислоты тэн – наиболее стойкое и наименее чувствительное к механическим воздействиям взрывчатое вещество [5, с. 131].
Возможность получения тэна из синтетического сырья позволила Германии в период второй мировой войны наладить его широкий промышленный выпуск. По данным немецкой трофейной документации известно, что производство тэна осуществлялось на пяти заводах, этерификацией пентаэритрита концентрированной азотной кислотой (96 98%) при температуре не выше 25°С (на двух заводах в непрерывном режиме, а на других периодическим способом). После второй мировой войны, судя по патенту Германии за 1969 год, сущность процесса не изменилась до сих пор.
В СССР разработка промышленной технологии производства тэна проводилась с 1929 по 1940 год НИИ-6 совместно с Чапаевским химзаводом. При разработке рационального способа производства тэна большое внимание было уделено его получению через сульфопентаэритрит (Ф.И. Блонштейн и др.). Этот способ представлялся в то время наиболее перспективным для промышленного использования, так как позволял использовать пентаэритрит низкого качества и не требовал высоколегированной стали для оборудования. Им же был разработан и непрерывный процесс, при полузаводской проверке которого было показано снижение расхода азотной кислоты на 40% по сравнению с периодическим. Однако на разделе фаз происходило образование и накопление малостойкой корки продукта, в связи с чем было принято решение проводить процесс нитрования периодически с систематической очисткой стенок аппарата от продукта. Периодическая технология была внедрена на Чапаевском химическом заводе в 1940 году. Несмотря на то, что в период войны 1941–1945 годы качество пентаэритрита оставалось недостаточно высоким, на заводе не наблюдалось аварийных случаев при нитровании сульфопентаэритрита.
Одновременно разрабатывались способы получения тэна прямым нитрованием пентаэритрита концентрированной азотной кислотой и серно-азотными смесями. Основным преимуществом этого способа была возможность применения аппаратуры из обычной стали. Однако по этому способу необходимо было использовать пентаэритрит только высокого качества, так как при работе на пентаэритрите с низкой температурой плавления получалась нестабильная реакционная масса.
В 1956 году К.Г. Костырев, С.С. Молчанова, Л.Ф. Николаева и др. показали, что способ нитрования пентаэритрита концентрированной азотной кислотой является более безопасным и позволяет получать тэн высокого качества из пентаэритрита с температурой плавления не ниже 230°С. В 1957 году на Чапаевском химзаводе и в 1966 году на Бийском олеумном заводе была внедрена технология производства тэна по периодическому способу этерификацией пентаэритрита концентрированной азотной кислотой с последующей перекристаллизацией полученного тэна из ацетона.
Химизм получения и области применения тэна
Пентаэритриттетранитрат (тэн) является азотнокислым, эфиром многоатомного спирта пентаэритрита. Впервые он был получен Толленсом в 1891 г. Из эфиров азотной кислоты это наиболее стойкое и сравнительно малочувствительное к механическим воздействиям взрывчатое вещество. Его формула:
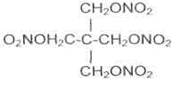
Тэн является одним из мощных бризантных взрывчатых веществ, для производства которого имеется практически неограниченная сырьевая база, так как первичными материалами для его получения являются синтетические продукты. Развитие органического синтеза позволило удешевить производство исходных продуктов для приготовления пентаэритрита – формальдегида и ацетальдегида, что и явилось стимулом для возникновения производства тэна. Формальдегид в настоящее время готовится в больших количествах из синтетического метанола. Ацетальдегид получается из ацетилена путем каталитической гидратации его в присутствии ртутных солей.
Все же в настоящее время стоимость тэна еще высока, и поэтому в мирной промышленности он применяется главным образом в капсюлях-детонаторах и идет для приготовления детонирующего шнура. Тэновые капсюли обладают значительно большей инициирующей способностью, чем гремучертутные и азидотетриловые.
Вследствие высокой чувствительности к механическим воздействиям тэн в чистом виде для снаряжения боеприпасов не применяется [13].
Тэн, или пентаэритриттетранитрат, является сложным азотнокислым эфиром четырехатомного спирта пентаэритрита.
Сам пентаэритрит С(СН2ОН)4 получается конденсацией ацетальдегида и формальдегида в присутствии гидроксида кальция по реакции:
2СН3СHO+8НСHO+Са(ОН)2=2С(СН2ОН)4+(НСОО)2Са.
Пентаэритрит представляет собой белое кристаллическое вещество с температурой плавления 260°С, не имеющее запаха и сладковатое на вкус. Он довольно хорошо растворим в воде (при 15°С в 100 частях воды растворяется 5,55 части пентаэритрита) [19, с. 79].
Пентаэритрит, особенно технический, содержит значительное количество примесей. Основная примесь дипентаэритрит – продукт конденсации двух молекул пентаэритрита:
2С(СН2ОН)4 →Н2О + С(СН2ОН)3 – СН2 – О – СН2 С(СН2ОН)3.
Дипентаэритрит, присутствуя в пентаэритрите, снижает температуру плавления последнего, образуя с ним при известных соотношениях (70% пента- и 30% дипентаэритрита), эвтектическую смесь, плавящуюся при 100°С. Дипентаэритрит плавится при 221°С. Технический пентаэритрит с температурой плавления 235°С содержит 10% дипентаэритрита.
Второй неизбежной примесью пентаэритрита являются сахаристые вещества, которые образуются вследствие альдольной конденсации формальдегида в щелочной среде.
6НСНО → СН2ОН(СНОН)4 – СНО.
Содержание этих примесей доходит до 0,26%.
Третьей примесью являются смолы, придающие продукту желтоватую окраску.
Из неорганических примесей основной является известь, которая обусловливает зольность продукта до 0,25%.
Большое содержание окисляющихся и сахаристых веществ в пентаэритрите вызывает во время получения азотокислого эфира побочные реакции, результатом которых является повышение температуры. Внезапное сильное повышение температуры при этерификации пентаэритрита азотной кислотой может вызвать вспышку и даже взрыв, поэтому необходимо тщательно следить за тем, чтобы содержание подобных примесей не превосходило норм, предусмотренных техническими условиями.
Качество пентаэритрита влияет также на выход тэна. Из пентаэритрита с температурой плавления 210°С выход кристаллизованного тэна составляет около 80%, а с температурой плавления 238°С – около 92%.
Пентаэритрит, применяемый для получения тэна, должен иметь температуру плавления не ниже 240°С и при опытной этерификации давать выход тэна не менее 90% от теоретического.
Действием азотной кислоты пентаэритрит легко может быть превращен в эфир по реакции:
С(СН2ОН)4 + 4HNO3= C(CH2ONO2)4 + 4Н2О.
При этом концентрация кислоты не должна быть ниже 86% HNO3. С повышением концентрации исходной азотной кислоты выход продукта увеличивается. Модуль ванны берется таким, чтобы отработанная кислота содержала 80–82% HNO3. Уменьшение модуля ванны и концентрации исходной азотной кислоты приводит к интенсивным окислительным процессам.
На окислительные процессы также влияют температура реакции и окислы азота. Поэтому температура этерификации не должна быть выше 20°С, а содержание окислов азота в азотной кислоте не более 2%.
Процесс этерификации пентаэритрита экзотермичен и протекает с большой скоростью, поэтому для поддержания указанной температуры необходимо очень энергичное перемешивание, хорошее охлаждение и порциальное прибавление спирта в кислоту.
По другому способу тэн получают через сернокислый эфир с последующей переэтерификацией при 55–60°С добавлением азотной кислоты.
При действии серной кислоты на пентаэритрит образуется сложный эфир пентаэритритсульфат по реакции
С(CH2OH)4+xH2SО4=C(CH2OH)4-X(CH2OSO3H)x+xH2O,
где х равен двум или трем, т.е. при действии серной кислоты получается пентаэритритдисульфат или пентаэритриттрисульфат [18, с. 334].
Превращение этого продукта в эфир азотной кислоты производится концентрированной азотной кислотой или меланжем по уравнению
C(CH2OH)4-x(CH2OSO3H)x+4HNO3= С(CH2ONO2)4 + (4-x) H2O+xH2SO4
Переэтерификация пентаэритритди- или пентаэритриттрисульфата происходит труднее, чем этерификация пентаэритрита; если этерификация последнего азотной кислотой идет с довольно большой скоростью даже при 0°С, то переэтерификация первого начинается при 35–40°С, а с достаточной скоростью протекает только при 55–60°С.
Промежуточным продуктом переэтерификации пентаэритритди- или пентаэритриттрисульфата является пентаэритритдинитратдисульфат, который не полностью превращается в тэн, а частично в нем остается.
Смешанные эфиры нестойки и являются причиной низкой стойкости нестабилизованного тэна. Для получения стойкого тэна, свободного от смешанных эфиров, этерификацию пентаэритритдисульфата азотной кислотой проводят при повышенной температуре порядка 55–60°С. Однако для полного удаления нестойких примесей тэн необходимо подвергать специальной стабилизации – кипячению с раствором соды (содовой варке) и перекристаллизации.
Промытый водой тэн обрабатывают в течение часа кипящим раствором соды, затем, после фильтрования, растворяют в ацетоне. В полученный раствор добавляют углекислый аммоний для нейтрализации оставшихся в тэне минеральных и, вероятно, органических кислот. Раствор отфильтровывают от избытка (NH4)2CO3 а также от других механических примесей, и для выделения кристаллов тэна охлаждают или выливают струей в двух-трехкратное количество воды.
Свойства тэна
Тэн представляет собой белое кристаллическое вещество с температурой плавления 141–142°С и удельным весом 1,77. Тэн плохо прессуется; прессованием можно достичь плотность 1,70 г./см. Теплоемкость тэна 0,4 кал/г°С [7, с. 224].
Тэн не гигроскопичен, растворимость его в воде при 19°С равна 0.01%, а при 100°С 0,035%.
Тэн вещество нейтральное и на металлы не действует. При продолжительном взаимодействии со щелочами и кислотами он разлагается.
Тэн достаточно стоек и превосходит по стойкости многие нитраты многоатомных спиртов. Свойство это объясняется тем, что четыре метоксильных группы располагаются вокруг центрального атома углерода; возможно, что существенное значение имеет также то обстоятельство, что тэн является твердым веществом.
Разложение тэна с выделением окислов азота при 140–145°С достигает значительной скорости уже через полчаса от начала опыта. При 175°С продукт интенсивно выделяет бурые пары окислов азота, а при 215°С происходит вспышка.
Воспламеняется тэн с трудом, зажженный (в небольших количествах) сгорает спокойно.
Тэн обладает высокой чувствительностью к удару: при падении груза в 2 кг с высоты 17 см он детонирует почти безотказно, но в отдельных случаях детонация происходит уже при высоте 15 см и даже при 10 см.
Основные взрывчатые свойства тэна:
Теплота взрывчатого разложения – 1385 ккал/кг
Объем газообразных продуктов взрыва – 790 л/кг
Скорость детонации – 8300 м/с
Расширение в бомбе Трауцля – 500 мл
Предельный вес детонатора:
Гремучая ртуть – 0,17 г.
Азид свинца 0,03 г.
Приведенные данные показывают, что тэн является мощным бризантным взрывчатым веществом.
Применяется он в качестве вторичного заряда в капсюлях-детонаторах для производства детонирующего шнура и для изготовления детонаторов артиллерийских взрывателей [1, с. 68].
Хорошими флегматизаторами тэна являются вазелин, парафин и церезин, но они существенно снижают его мощность.
Требования, предъявляемые к кристаллизованному тэну, следующие:
1) внешний вид – мелкокристаллический порошок белого цвета (допускается слабо-серый оттенок) без посторонних примесей, видимых на глаз, и без явных признаков подмочки;
2) температура плавления в пределах 138–140°С;
3) содержание влаги и летучих веществ не более 0,1%;
4) содержание нерастворимых в ацетоне при обыкновенной температуре примесей не более 0,1%;
5) зольность не более 0,2%, в том числе кремнезема не более 0,01%;
6) отсутствие свободных кислот;
7) стойкость, определенная по концентрации водородных ионов при 110°С в течение 8 часов, не ниже 5,5;
8) стойкость по иодокрахмальной пробе 1 час при 80°С.
Технология производства тэна
Особенностью процесса производства тэна является то, что исходный продукт пентаэритрит представляет собой твердое вещество с высокой температурой плавления. Дозировка твердого исходного компонента значительно труднее, чем дозировка жидкостей [3].
Промышленное производство тэна может быть осуществлено двумя способами: двухстадийным – с предварительным получением сульфата пентаэритрита и последующим превращением его в нитрат, и одностадийным – непосредственным получением нитрата пентаэритрита.
И в том и в другом случаях процесс осуществляют путем добавления в соответствующий аппарат, наполненный серной или азотной кислотой, пентаэритрита. При этом последний растворяется в указанных кислотах. Процесс растворения предшествует реакции этерификации и, по-видимому, задает общую скорость.
Хорошее перемешивание и наличие достаточно мелкого (не слипшегося в комочки) пентаэритрита является необходимым условием технологического оформления процесса этерификации.
Двухстадийный способ
Этот способ был предложен раньше одностадийного, так как, казалось, имел больше перспектив для внедрения. Процесс состоял из двух стадий: первая – получение сернокислого эфира пентаэритрита и вторая – получение азотнокислого эфира пентаэритрита. Как первую, так и вторую стадии можно вести при повышенной температуре (50–60°С), не опасаясь окисления даже во второй стадии, так как при получении нитрата гидроксильные группы защищены, и, кроме того, окислы азота, вызывающие этот процесс, связываются серной кислотой. Повышенная температура, необходимая для замены сульфогруппы нитрогруппой, позволяет применить для охлаждения речную воду.
Этерификацию серно-азотной смесью можно проводить в аппаратуре из обычной стали. Одностадийный же способ получения тэна требует наличия холодильной установки и аппаратуры из легированной стали.
Предполагалось, что двухстадийный способ производства тэна значительно легче осуществить в аппаратуре непрерывного действия, чем одностадийный, так как в этом случае в аппарат будет дозироваться не пентаэритрит, а раствор его сульфоэфира в серной кислоте. Непрерывная дозировка жидких компонентов, безусловно, проще и точнее, чем твердых.
Описана следующая схема получения тэна по двухстадийному способу. В первой стадии готовится 10–15%-ный раствор пентаэритрита в серной кислоте. Во второй стадии этот раствор одновременно с азотной кислотой вводят в аппарат предварительного смешения, снабженный мешалкой и охлаждающей поверхностью, где поддерживается температура 12°С. Смесь передается в нижнюю часть второго аппарата, представляющего собой колонну (с рубашкой, в которой циркулирует вода с температурой 40–50°С). Реакционная масса в колонне нагревается до 55–60°С, поднимается вверх и вытекает через верхний штуцер. Скорость движения жидкости в колонне регулируется таким образом, чтобы процесс переэтерификации успел полностью закончиться до момента выхода из аппарата [18, с. 339].
Осуществить двухстадийный способ получения тэна в аппаратуре непрерывного действия трудно из-за низкой стойкости продукта.
Тэн, получаемый этим способом, в неочищенном виде имеет чрезвычайно низкую стойкость. Какая-либо задержка такого тэна в аппарате, например, налипание на внутренней стороне крышки или стенки, а также на змеевиках, приводит к саморазложению продукта, которое может закончиться взрывом. Во избежание указанного необходима тщательная промывка аппаратуры.
Тэн, полученный из сульфата пентаэритрита, нуждается в специальной стабилизации. Нестабилизованный тэн имеет склонность к разложению даже при обыкновенной температуре.
Одностадийный способ
При этом способе технологический процесс разбивается на ряд операций:
1) Cушка, измельчение и просеивание пентаэритрита;
2) получение тэна;
3) промывка тэна;
4) кристаллизация тэна;
5) сушка тэна;
6) просеивание тэна и его укупорка.
Пентаэритрит, идущий на изготовление тэна, предварительно измельчают, сушат и просеивают. Сушка пентаэритрита обычно производится в барабанной сушилке при температуре не выше 100°C [25].
Получение тэна проводят по схеме, изображенной на рис. 1 (см. приложение).
В нитратор 1 из мерника 2 вливают 300 кг азотной кислоты (93-95%) и постепенно при работающей мешалке засыпают 60 кг пентаэритрита с такой скоростью, чтобы температура не поднималась выше 20°С. После введения всего пентаэритрита дается выдержка 30 мин. В процессе этерификации происходит образование тэна, который выпадает из отработанной кислоты (уд. вес 1,43) в виде кристаллов.
Реакционная масса из нитратора спускается на вакуум-воронку 4, где тэн отжимается от отработанной кислоты, собираемой в вакуум-сборник 6. Затем кислый тэн переносят для предварительной промывки в разбавитель 7, наполненный водой.
Разбавитель представляет собой бак, снабженный мешалкой и ложным дном из пористой керамики. Мешалка необходима для предотвращения местных перегревов в момент погружения кислого тэна в воду. На 1 часть тэна в разбавитель предварительно заливают 6,5 части воды.
Кислый тэн перемешивают с водой 15 мин, затем мешалку останавливают и посредством вакуума кислую воду отжимают через ложное дно разбавителя. Затем здесь же в разбавителе тэн еще три раза промывают полуторакратным количеством воды. После такой промывки кислотность тэна составляет около 1%.
Затем тэн направляют на окончательную промывку, которая производится следующим образом.
В промывочный чан загружают 1%-ный раствор соды (в 8–10-кратном количестве по отношению к тэну), нагревают до 90°С и затем постепенно вносят тэн, при этом происходит вспенивание массы за счет выделяющейся углекислоты. После загрузки массу перемешивают в течение часа при температуре 85–90°С. Во все время промывки среда должна оставаться щелочной. В процессе промывки комочки кристаллов тэна заметно рассыпаются и кислота, находящаяся между ними, нейтрализуется содой.
После содовой варки тэн отжимают от воды на вакуум-воронке. Полученный сырой тэн имеет кислотность около 0,5–0,8%. Достичь кислотности меньшей 0,3% многократной промывкой тэна не удается, по-видимому, вследствие наличия внутрикристальной кислоты. Для удаления этой кислоты тэн подвергают перекристаллизации из ацетона. С этой целью тэн растворяют в ацетоне, для нейтрализации кислоты добавляют углекислый аммоний и некоторое время кипятят раствор. Далее тэн осаждают из раствора либо охлаждением, либо разбавлением раствора водой.
Так как тэн, поступающий на стабилизацию, содержит 15–18% воды, которая снижает его растворимость, то для растворения на 1 часть его приходится брать избыток ацетона (2,2–2,5 вес. ч.). В полученный раствор присыпают мелкоизмельченный углекислый аммоний из расчета 100% избытка против необходимого для нейтрализации кислотности тэна. Растворение тэна и стабилизацию его производят при 58°С в течение часа. По истечении этого времени раствор передавливается через фильтр в кристаллизатор, предварительно нагретый до 50°С.
В холодный аппарат спускать горячий ацетоновый раствор тэна нельзя во избежание выкристаллизовывания продукта на стенках и в трубах. Поэтому раствор медленно охлаждают водой, подаваемой за рубашку. Если охлаждение быстрое, то образуется корка, которую растворяют нагреванием аппарата до 58°С [24, с. 87].
По окончании кристаллизации тэн отжимают от ацетона на вакуум-воронке. После этого тэн содержит 15–20% маточного ацетона, который удаляют промывкой спиртом.
Ацетон используют три раза, добавляя каждый раз в маточный ацетон свежий. Отработанный ацетон идет на ректификацию.
Отжатый тэн, содержащий 15–20% спирта, поступает в камерные сушилки, где его сушат при 40°С в течение 12 час. Высушенный тэн просеивают и укупоривают в миткалевые мешки.
Этот способ производства тэна применялся во время второй мировой войны в Германии, где его осуществляли в аппаратуре непрерывного действия. В Германии имелось две схемы производства тэна, в которых по-разному производились отжим и стабилизация отработанной кислоты.
1) Непрерывный способ получения тэна с непрерывным отжимом отработанной кислоты показан на схеме (см. приложение рис. 2).
В нитраторы 2 (емкостью 50 л каждый) при перемешивании непрерывно через дозаторы подаются пентаэритрит и азотная кислота (97–98% HNO3) в соотношении 1: 5 и при температуре 10–25°С. Из нитраторов 2 реакционная масса непрерывно поступает в V-образный буферный нитратор 3 (емкость 40 л), в котором масса охлаждается до 10–15°С.
Из буферного нитратора реакционная смесь непрерывно поступает на вакуум-фильтр 4 (вращающийся со скоростью 0,75 об/мин и имеющий фильтрующую поверхность 0,5 м). На фильтре тэн отжимается до 12 – 15% кислотности и непрерывно поступает в промывную колонну 5, где промывается холодной водой. Разгрузка промывной колонны производится периодически на вакуум-воронку 7. Здесь тэн дважды промывается горячей и один раз холодной водой, отжимается до 12–15% влажности. Промытый тэн выгружают вручную в мешки и направляют на кристаллизацию.
Промывные воды, содержащие 8–12% HNO3, спускают через ловушки в канализацию. Отработанная кислота поступает в алюминиевые отстойники 9 на 24 часа, затем сливается в сборник 10, а из него передается в хранилище отработанной кислоты. Состав отработанной кислоты следующий: 80–82% HNO3; 0,5–0,75% N2O3 и 1,0–1,5% нитровеществ. После отстоя отработанная кислота содержит нитровеществ около 0,75%.
Выделяющиеся нитрозные газы поглощаются водой в абсорбционной колонне и после концентрирования полученной в колонне кислоты возвращаются в производство.
Отработанная кислота для стабилизации направляется после отстоя в так называемую цершторколонну, состоящую из пяти последовательно соединенных колонн, снабженных рубашкой для подогрева паром до 90 – 100°С. Пройдя последовательно все пять колонн, кислота освобождается от нитропродукта и поступает в холодильник, где охлаждается до 25–30°С. Основная масса нитровеществ разлагается в первых двух колоннах. Для усиления разложения нитровеществ в третью колонну подается вода, в результате чего выделяющиеся при этом окислы азота усиливают процесс кисления. Отработанная кислота после стабилизации содержит 65–70% NO3 и свободна от нитровеществ [18, с. 342].
Кристаллизация тэна (см. приложение рис. 3) проводится в отдельном здании, в котором размещены два аппарата для растворения, емкостью по 600 л и два кристаллизатора емкостью по 1100 л.
Аппараты для растворения и кристаллизаторы изготовлены из алюминия и снабжены мешалками, делающими 150 об/мин, и обратными холодильниками.
Ацетон в количестве 360 л из хранилища 1 сжатым воздухом (или азотом) подается в автоматический мерник 2, а из него через предохранительный горшок в подогреватель 3, где нагревается до 50°С; затем он поступает в растворитель 4, куда загружают 112 кг влажного (100 кг сухого) тэна и 750 г. бикарбоната натрия. После растворения тэна (30 мин) раствор спускают через фильтр 5 в кристаллизатор 6, где к раствору добавляют еще 750 г. бикарбоната натрия или аммония. Бикарбонат натрия добавляют в два приема, чтобы не создавать сильно щелочной среды, которая вызывает коррозию алюминиевой аппаратуры.
К ацетоновому раствору в кристаллизатор постепенно приливают из мерника 7 600 л холодной воды со скоростью 30 л/мин при работающей мешалке. При таком режиме осаждения получается тэн с хорошей сыпучестью.
Из кристаллизатора ацетон, разбавленный до 30% с выделившимся тэном, спускают на вакуум-воронку 8, тэн отжимают и промывают дважды теплой и один раз холодной водой. Отжатый от воды тэн (с влажностью 1О – 15%) выгружают в мешки и отправляют либо на сушку, либо на флегматизацию.
Флегматизация производится искусственным воском. Во флегматизатор загружают тэн и воду (1:2) и при перемешивании подают расплавленный воск. Температуру во флегматизаторе поддерживают на 4–5°С ниже температуры плавления воска. Затем температуру во флегматизаторе поднимают на 2–3°С выше температуры плавления воска, и после 15–20 мин перемешивания содержимое флегматизатора охлаждают и фильтруют.
Флегматизированный тэн с целью получения большой однородной партии (1000 кг) смешивают под водой и после вторичной фильтрации направляют на сушку.
Готовый тэн должен иметь температуру плавления не ниже 138°С, содержать азота 17,4% и нерастворимых в ацетоне веществ не больше 0,3%.
Разбавленный ацетон поступает на ректификацию в ректификационные колонки Гольцен-Гримма (до концентрации 98%), после чего его снова используют на кристаллизацию.
Этот способ получения тэна является достаточно простым, сравнительно безопасным, высокопроизводительным и обеспечивает большой выход и высокое качество готового продукта при относительно низких расходных коэффициентах.
2) Непрерывный способ с периодическим отжимом отработанной кислоты (см. приложение рис. 4) [18, с. 343].
Пентаэритрит через воронку и автоматические весы посредством шнека дозируется в основной нитратор (объем 0,5 м), куда одновременно из хранилища через дозатор подается 99%-ная азотная кислота. На 1 вес. ч. пентазритрита дозируется 5–6 вес. ч. азотной кислоты. Температуру в нитраторе, равную 15°С, поддерживают с помощью рассола, охлажденного до 11°С.
Нитромасса из основного нитратора перетекает в буферный нитратор (объем 0,17 м), а затем в разбавитель (объемом 0,17 м), куда дозируется вода. Количество воды должно быть таким, чтобы концентрация отработанной кислоты снизилась с 80 до 40% по HNO3. Температуру в буферном нитраторе поддерживают равной 10°С, а в разбавителе 15°С (охлаждают также рассолом). При разбавлении тэн выкристаллизовывается, и всю массу подают на вакуум-фильтр.
Вакуум-фильтр в центре фильтрующего полотна имеет отверстие, к которому подведена труба, соединяющая его с баком для промывки. Во время загрузки и отжима отверстие закрыто втулкой. После отсоса кислоты втулку вынимают, и кристаллы тэна смывают в бак для промывки. Из бака для промывки массу передают на фильтр, и после отжима кислой воды тэн мощной струей воды смывают в бак для нейтрализации кислого тэна раствором соды. Обработанный содой тэн спускают на фильтр, и после отжима передают на установку непрерывной очистки.
В растворителе, куда тэн подают одновременно с ацетоном, происходит полное его растворение. Раствор профильтровывается и перетекает в бак для разбавления водой. При разбавлении раствора тэн выкристаллизовывается. Всю массу пропускают через дистилляторы для удаления ацетона. По выходе из последнего дистиллятора горячую массу охлаждают и фильтруют. При получении флегматизированного тэна в третий дистиллятор вводят расплавленный монтан-воск (воск из бурого угля).
Расположены нитраторы в отдельном помещении, валы мешалок выведены через потолок. Установка оснащена контрольно-измерительными приборами, все управление ею автоматизировано.
Рядом с нитрационным помещением размещается холодильная установка и баки для хранения кислоты. Здесь же расположено здание для обработки сырого тэна содой.
После промывки тэн передают в следующее здание для перекристаллизации и флегматизации.
Потери ацетона при периодической перекристаллизации составляют 0,125 т на одну тонну готового тэна, а при непрерывной – 0,055 т.
Отработанную кислоту подвергают отстаиванию и затем концентрируют под вакуумом до 98–99%.
1.3 Гексоген: свойства и технология производства
Впервые гексоген (циклотриметилентринитрамин) был получен в 1897 году, а в 1898 году Гениннг запатентовал способ получения через динитрат уротропина. В 1920 году Герц получил гексоген непосредственным нитрованием уротропина азотной кислотой (окислительный способ) и показал, что он является взрывчатым веществом. С этого времени гексоген рассматривается как мощное бризантное взрывчатое вещество [5, с. 123].
В настоящее время взрывчатые смеси на основе гексогена широко применяются для снаряжения боеприпасов во многих странах.
В Англии и других странах получение гексогена по окислительному способу Герца на установках непрерывного действия началось уже в 1932–1933 годах. Во время второй мировой войны был разработан еще ряд способов, а производство гексогена мощностью около 350 тонн в сутки действовало в Англии, США и Германии. В 1942 году Англией совместно с Канадой и США был разработан более выгодный с экономической точки зрения уксусно-ангидридный метод.
Первые работы по получению гексогена в СССР начинаются в 1929–1930 годах в НИИ-6 сотрудниками И.А. Сыркиным, Н.И. Быстровым, А.А. Гринбергом. За основу способа получения был принят способ Герца. Уточнялись факторы, влияющие на повышение выхода продукта, исследовалось влияние окислов азота на стойкость нитромассы, обеспечивавшей безопасность ведения технологического процесса. Одновременно эти же авторы исследовали способ получения гексогена через динитрат уротропина, который позволял увеличить выход продукта, был более безопасным вследствие распределения выделяемой в процессе реакции теплоты по стадиям. Однако, несмотря на преимущества, они были вынуждены отказаться от промышленного использования способа, в связи с многостадийностью и сложностями техпроцесса.
Таким образом, к 1932 году для проверки остался только способ получения гексогена прямым нитрованием уротропина азотной кислотой, который в 1933 году впервые был проверен на Чапаевском химзаводе на опытной установке в периодическом варианте. Проверка процесса на опытной полузаводской установке подтвердила правильность выбранных в лабораторных условиях технологических параметров нитрования. Однако процесс кристаллизации из кислоты разбавлением нитромассы не был безопасным. Отработанные кислоты из-за наличия примесей были нестойкими, что затрудняло их хранение и переработку.
В этот же период НИИ-6 была проведена оценка стойкости гексогена, высаженного из нитромассы, и было показано, что такой продукт является достаточно стойким и может использоваться для снаряжения боеприпасов. Для гексогена. применяемого в капсюлях-детонаторах, по требованию военных была введена дополнительная перекристаллизация из ацетона.
В 1936 году там же начинается разработка технологии получения гексогена непрерывным способом (З.В. Владимирова и А.А. Гринберг), который в 1938 году проверяется на опытной установке завода им. Я.М. Свердлова (Е.М. Адаскин, М.А. Цесарская и Е.М. Прилуцкий). Технологические параметры процесса нитрования, предложенные авторами, до настоящего времени практически остались неизменными. Высаживание гексогена из нитромассы велось методом непрерывного разбавления водой в аппарате-кристаллизаторе. Гексоген отделялся от отработанной кислоты на вакуумных воронках, а отработанная кислота сбрасывалась в канализацию. Удаление газов из аппаратов проводилось с помощью эжекторов. Проверка процесса получения гексогена непрерывным способом на опытной установке показала возможность организации непрерывного процесса нитрования. При отработке было обнаружено неоднократное загорание уротропина в нитраторе, воронке и питателе. Однако это явление не удавалась исключить в течение многих лет. Были также случаи выброса массы из кристаллизатора. Причина заключалась в том, что, после разбавления нитромассы водой и выделения гексогена, в отработанной кислоте оставались нестойкие примеси, разложение которых сопровождалось большим газовыделением, поэтому перед исследователями встала задача по стабилизации отработанной кислоты.
При выборе варианта для реализации в промышленности в первую очередь учитывались вопросы безопасности, экономической и технологической целесообразности. Были рассмотрены три способа получения гексогена, предложенные НИИ-6: нитрация уротропина с утилизацией отработанной кислоты в виде аммиачной селитры; получение через динитрат уротропина с регенерацией формальдегида и нитрация уротропина концентрированной азотной кислотой. Последний способ был признан наиболее приемлемым, т.к. имел наименьшее количество технологических стадий, позволял обеспечить возврат отработанной кислоты после регенерации в технологический цикл, что приобретало особую важность в период военного времени, и был рекомендован для проектирования.
В 1940 году ГСПИ приступает к проектированию производства гексогена. Одновременно на заводе им. Я.М. Свердлова начинаются работы по строительству цеха, а в апреле 1942 года начался выпуск гексогена. Война расставила свои акценты и потребовала ввода дополнительных мощностей по производству гексогена. В декабре 1941 года Государственный комитет обороны обязал Наркомат боеприпасов и Наркомхимпром приступить к немедленной организации новых производств гексогена. В Кемерово в приспособленных зданиях была освоена первая очередь периодического производства гексогена, а 20 января 1942 года была получена и поставлена заказчику первая партия гексогена. В августе 1942 года на этом же заводе вводится в эксплуатацию вторая очередь производства гексогена.
Свойства гексогена
Гексоген представляет собой белые кристаллы без запаха и вкуса, он является сильным ядом, поэтому при работе с ним необходимо строго соблюдать правила техники безопасности [23, с. 58].
Удельный вес гексогена – 1,816, гравиметрическая плотность 0,8 – 0,9 кг/л. Прессованием под давлением 2000 кг/см2 достигается плотность 1,73. Температура плавления 204,5–205°С. Технический продукт, полученный прямым нитролизом уротропина азотной кислотой, плавится при 202°С, что соответствует содержанию примесей около 1%. Теплоемкость гексогена 0,30 кал/г°С, теплота кристаллизации 21,3 ккал / моль, его формула:
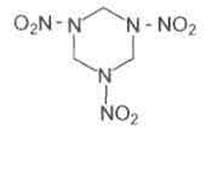
Гексоген не гигроскопичен, плохо растворим в воде, эфире, спирте, хлороформе и слабой азотной кислоте. Хорошо растворим в ацетоне и концентрированной азотной кислоте.
К действию солнечного света гексоген стоек.
Гексоген – вещество нейтральное, с разбавленными кислотами не реагирует.
В концентрированной азотной кислоте на холоду гексоген растворяется без разложения и может быть осажден из нее простым разбавлением кислоты водой.
При обработке гексогена раствором щелочи в водном растворе ацетона происходит гидролиз. Энергия активации этого гидролиза равна 14 ккал / моль. Большая скорость гидролиза гексогена разбавленным раствором каустической соды используется в производстве для очистки аппаратов от гексогена [11, с. 93].
Гексоген, приготовленный уксусноангидридным способом, содержит в качестве примеси октоген.
Гексоген обладает высокой стойкость, он может месяцами храниться при 50°С без разложения и выдерживает пробу Абеля при 60°С свыше 60 часов. Стойкость химически чистого гексогена значительно превышает стойкость тетрила, но стойкость технического продукта нередко ниже. Причин недостаточной стойкости технического гексогена может быть две.
Первой является наличие в продукте примеси диацетокситетраметилентринитроамина (BSX). Последний может находиться в гексогене, полученном уксусноангидридным методом. Гексоген стабилизируют длительным кипячением в воде, во время которого нестойкий диацетокситетраметилентринитроамин разлагается.
Второй причиной недостаточной стойкости технического гексогена считают наличие в нем неотмытой азотной кислоты. Этим недостатком обладает гексоген, полученный нитролизным методом, при котором гексоген выделяется из 55%-ной азотной кислоты. Образующиеся кристаллы гексогена содержат связанную азотную кислоту, удаление которой происходит очень медленно даже при длительном кипячении и при наличии мелких кристаллов. Для получения 0,13% кислотности гексогена требуется длительное время промывки (40–50 часов), что в условиях производства трудно достижимо. Наличие в техническом гексогене значительного количества азотной кислоты вызывает коррозию и затрудняет использование стандартных проб при определении стойкости. Показано, азотная кислота практически не снижает стойкости гексогена. Таким образом, вторая причина низкой стойкости технического гексогена является по существу кажущейся.
Гексоген при нагревании начинает разлагаться с заметной скоростью при 200°С. Температура его вспышки равна 230°С. На открытом воздухе гексоген сгорает ярким белым пламенем без остатка, при быстром нагревании разлагается со взрывом [19, с. 44].
Чистый гексоген термически стоек.
По чувствительности к удару гексоген занимает среднее место между тетрилом и тэном; груз в 2 кг вызывает взрыв гексогена при падении с высоты 30–32 см. Чувствительность по стандартной пробе (р=10 кг, h = 25 см) – 80%.
Теплота взрывчатого разложения гексогена 1320 ккал/кг, объем газообразных продуктов взрыва 910 л/кг, скорость детонации (р=1,7) 8400 м/с, расширение в бомбе Трауцля 470 мл.
Взрывчатые свойства флегматизированного гексогена значительно снижаются по мере увеличения количества флегматизатора.
Гексоген применяется для снаряжения снарядов малого калибра, кумулятивных зарядов, в детонаторах, в капсюлях-детонаторах. В смеси с алюминиевой пудрой или с тротилом его используют для снаряжения различных боеприпасов.
Гексоген используют также в так называемых пластичных ВВ или во взрывчатых замазках. Смеси из гексогена и связывающего материала являются мягкими, пластичными и немного клейкими. Они применяются для подрывных целей, например, с их помощью можно резать металл, мостовые фермы, танки, доты и т.п. Подобные смеси состоят из 88 частей тонкоизмельченного гексогена и 12 частей смазочного масла или из 78% гексогена и 22% смолистого связывающего вещества из нитропроизводного ароматического углеводорода и нитроцеллюлозы [7, с. 223].
Технология производства гексогена
Получение гексогена окислительным методом
Английский вариант
Технологический процесс получения гексогена состоит из трех стадий: нитрования, промывки и сушки, проводимых в отдельных зданиях.
Нитрование. На этой стадии производят нитролиз уротропина путем непрерывной дозировки кристаллического уротропина и концентрированной азотной кислоты в охлажденную реакционную смесь из тех же ингредиентов. Получающийся раствор содержит гексоген и различные нежелательные побочные продукты, а также значительное количество неиспользованной азотной кислоты.
Этот раствор перетекает в сосуд, в котором производится разбавление его водой для выкристаллизовывания гексогена. Поддерживая высокую температуру нитромассы, дают возможность произойти распаду растворенных в ней побочных продуктов. Остаточную слабую кислоту отделяют от твердого гексогена путем непрерывного фильтрования и смешивают перед отправкой на концентрацию со слабой кислотой, полученной из абсорбционных колонн.
Указанные процессы проводят в параллельно расположенных двух или трех нитраторах и двух разбавителях.
Нитратор представляет собой прямоугольный сосуд из нержавеющей стали, разделенный перегородками на три отделения, снабженные мешалками. В первом отделении установлено три концентрически расположенных змеевика (поверхность охлаждения 1,8 м). Во втором и третьем отделениях установлено по одному змеевику. В первой и второй секциях температура нитромассы должна быть не выше 25°С. В третьей секции нитратора в змеевике циркулирует горячая вода и температура нитромассы поддерживается на уровне 38°С, что способствует увеличению выхода гексогена.
От крышки нитратора отведены две трубы для газов, причем в первой имеется цилиндрическое смотровое окошко из стекла «пирекс», через которое наблюдают за цветом отходящих газов. Появление значительного количества окислов азота сигнализирует о необходимости немедленного спуска содержимого нитратора в аварийный чан. В крышке над первым и вторым отделениями имеются отверстия, через которые производится подача уротропина. В первом отделении установлен питательный аппарат для подачи азотной кислоты. Спускное отверстие находится на стенке третьего отделения.
В дне нитратора вмонтирован спускной клапан, через который содержимое нитратора стекает в бак с водным раствором мочевины. При сливе нитромассы в этот раствор выделяется значительно меньше газов, чем при сливе в чистую воду.
Нитромасса из нитратора поступает в стояк, питающий разбавитель, который расположен несколько ниже.
Разбавители по своей форме сходны с нитраторами, но значительно больше по размерам. Рабочая глубина составляет 0,7 м. В каждом разбавителе имеется четыре камеры для размешивания, снабженные охлаждающими змеевиками [18, с. 276].
Мешалки имеют плоские лопасти и работают при 195 об/мин.
Нитромасса выходит из разбавителей через закрытый желоб и поступает в охладник, состоящий из трех камер (основной и двух дополнительных). Холодная масса, перетекающая из этих камер, поступает на фильтры-классификаторы.
В начале нитрования разбавитель наполняют 55%-ной азотной кислотой, последняя нагревается примерно до 100°С паром, циркулирующим в змеевиках. Подачу пара постепенно уменьшают по мере поступления нитромассы из нитратора и включают подачу воды. Воду дают в таком количестве, чтобы концентрация выходящей кислоты была 55%. В разбавителе поддерживают температуру 75°С.
Нитромасса поступает в первую камеру, в которую через крышку по трубе подается вода для разбавления. Для этой цели используют промывную воду. Количество подаваемой воды регулируется через ротаметр.
Нитрационное здание имеет пульт управления.
Масса сырого гексогена и слабая азотная кислота поступают из охладника по закрытому желобу на первый фильтр, где отработанная кислота отделяется от гексогена. Затем гексоген попадает во второй фильтр, в котором промывается водой для удаления оставшейся кислоты. Кислая вода засасывается в отстойник и в дальнейшем ее используют для разбавления нитромассы [6].
Сырой гексоген, выходящий из второго фильтра, попадает в вагонетку (из нержавеющей стали), в которой его переводят в промывное отделение. На другом заводе гексоген с фильтра смывается водой в деревянный чан, снабженный мешалкой (500 об/мин). Суспензию гексогена перекачивают из чана в промывочное отделение по трубе диаметром 50 мм с помощью пароструйного эжектора.
Промывка. Сырой гексоген содержит часть крупнозернистых частиц, которые необходимо измельчать, так как в них (в промежутках между кристаллами) содержится азотная кислота (0,1–0,2%). Кроме того, азотная кислота находится и в смывной жидкости, остающейся на сыром гексогене. Назначением технологических процессов, проводимых в этой стадии, является измельчение крупных зерен гексогена и удаление (путем промывки) азотной кислоты.
Вагонетку с сырым гексогеном, поступившую в здание промывки, подкатывают под пароструйный прибор, к всасывающему концу которого прикреплен армированный шланг. Воду добавляют к массе гексогена при помощи водяного рукава; вся масса всасывается паровым эжектором по мере смачивания гексогена. Таким путем одна работница может опорожнить и наполнить вагонетку в течение пяти минут.
Суспензия эжектируется паром и поступает в один из двух мешателей из нержавеющей стали с закругленным днищем диаметром 1,2 м и высотой 2,1 м. Из этих приемников масса воды и гексогена в соотношении примерно 3: 1 подается самотеком на вальцы, где разрушаются гранулы. Одновременно с массой гексогена на вальцы подается вода для предотвращения нагревания в случае нарушения подачи. Измельченная масса поднимается над вальцами через трубу диаметром 25 мм из нержавеющей стали примерно на высоту 460 мм и может быть направлена в любой чан, при помощи резинового шланга.
В чан загружают около 2000 кг гексогена. После поступления суспензии гексогена с водой в чан мешалку останавливают, дают суспензии отстояться в течение 45 мин, затем через клапан для декантирования сливают кислую воду в деревянную ловушку. Трижды промывают массу холодной водой и после подачи воды для четвертой промывки нагревают массу острым паром до 90–100°С и выдерживают при этой температуре в течение 12 час. [18, с. 278].
После декантации влажный гексоген спускают из чана и направляют для дальнейшего использования.
Сушка. Сушат гексоген в вакуум-сушильных шкафах при 60°С.
Гексоген, предназначенный для приготовления смесей с тротилом, не сушат: удаление влаги происходит во время смешения его с расплавленным тротилом.
Количество исходных продуктов на одну тонну гексогена (в т):
| Уротропин | 0,83 |
| Азотная кислота | 8,87 |
| Из этого количества: | |
|
возвращается в виде отработанной кислоты (55%-ной HNO3) |
3,48 |
| улавливается абсорбционной системой | 3,43 |
| потребляется на нитролиз | 1,87 |
|
Для концентрации отработанной кислоты расходуется H2SO4 |
0,49 |
Следовательно, выход гексогена составляет по формальдегиду 37,7%, по аминогруппам – 56,7%, по азотной кислоте – 9,7%. Концентрация используемой азотной кислоты 95–95%.
Немецкий вариант
Метод «SH» разработан в 1937 г. Шнурре.
В отличие от английского варианта процесс проводят в аппаратах периодического действия, используя для нитролиза уротропина меньшее количество азотной кислоты, но более высокой концентрации (99% HNO3).
Схема технологического процесса получения гексогена изображена на рис. 5 (см. приложение).
В нитратор 2 загружают азотную кислоту, охлажденную до 0°С, и к ней медленно присыпают предварительно высушенный уротропин, поддерживая температуру не выше 20°С. Во избежание распиливания уротропин рекомендуется подавать в виде спрессованных шариков. После окончания нитрования содержимое нитратора спускают в буферный аппарат 3, из которого нитромасса последовательно проходит по аппаратам 4, 5, 6, 7 и 8, где уротропин полностью донитровывается. В буферных аппаратах с третьего по восьмой поддерживается температура 15–20°С. Далее нитромасса из буферного аппарата 8 идет для стабилизации в два окислителя (основной 9 и буферный 10), куда одновременно подается вода для разбавления. В окислителях поддерживается температура 70–80°С [10, с. 61].
Затем суспензия гексогена в отработанной кислоте перетекает в охладник 11 и последовательно проходит также еще через три охладника 12, 13, 14. В первых трех охладниках происходит естественное охлаждение, в четвертом нитромасса охлаждается водой до 20°С.
Из охладника 14 нитромассу спускают на барабанный фильтр (или центрифугу) 15, отработанную кислоту спускают в приемник 16, а гексоген промывают два-три раза холодной водой, затем смывают водой в мешатель 17. В мешателе гексоген размешивают с водой и суспензию центробежным насосом качают в автоклавы 18 (давление в автоклавах 2,5–3 атм.), расположенные в другом здании, где нагревают до 130–140°С и при этой температуре выдерживают 4–5 час.
По окончании варки в автоклаве массу охлаждают до 100° и спускают в аппараты для флегматизации 19, где находится расплавленный искусственный воск, подкрашенный Суданом. Воск берут из расчета содержания его в гексогене 5%. После слива суспензии гексогена к воску массу резко охлаждают до 20°С в течение 30 мин и спускают на фильтр-воронки 20, где гексоген отжимают в течение двух часов до 10% влажности. Отжатый гексоген выгружают в шелковые мешки и отправляют на другой завод на сушку.
Кислотность гексогена после промывки на воронке 0,3–0,4%, а после варки в автоклаве 0,1%. Температура плавления полученного гексогена 202°С. На одну тонну гексогена расходуется: уротропина 0,83–0,84 г. и азотной кислоты (99%-ной) 7,1 т, из которой регенерируется 5,2 т [24, с. 67].
Окислительный метод получения гексогена, дающий продукт высокого качества, весьма прост, надежен и достаточно безопасен, однако обладает рядом существенных недостатков. Главными из них являются: большой расход концентрированной азотной кислоты и малый выход гексогена по формальдегиду (35–40% от теоретического). Необходимость двойного разбавления кислоты водой с целью выделения гексогена также резко удорожает концентрацию отработанной кислоты для повторного ее использования. Учитывая эти недостатки, исследователи ряда стран вели работы по разработке более совершенных методов получения гексогена. В Германии во время второй мировой войны гексоген получали и другими методами: «уксусноангидридным» («КА»), через «белую соль» («W»), конденсацией формальдегида с аммонийной селитрой в присутствии BF3 («E»).
Получение гексогена методом «W»
Во время второй мировой войны в Германии работала опытная установка, на которой производили гексоген весьма оригинальным методом «W» (через сульфаминовую кислоту). Принципиальная схема установки показана на рис. 6 (см. приложение).
В конденсаторе 1 происходит взаимодействие аммиака с серным ангидридом, которые дозируют в соотношении 1:3, при этом образуются иминосульфонат и иминодисульфонат аммония:
SО3 + 2NH3 = NH2SО3NH4;
2SО3 + 3NH3 = NH (SO3NH4)2.
Полученную соль растворяют в трехкратном количестве воды и спускают в чан 2, затем добавляют оксид кальция в количестве 110% от теоретически необходимого. Раствор нагревают острым паром для удаления аммиака и отфильтровывают на воронке 3 от избытка гидроксида кальция. Фильтрат принимают в чан 4, где подкисляют серной кислотой, подаваемой из мерника 5. При этих операциях протекают следующие реакции:
NH2SО3NH4+NH(SО3NH4)2 + 2Са(ОН)2 = (NH2SО3)2Ca + CaSО4 + 3NH4OH.
При подкислении иминосульфонат кальция гидролизуется с образованием сульфаминовой кислоты.
Для получения калиевой соли сульфаминовой кислоты в чан 4 после подкисления и перемешивания добавляют оксид кальция и бисульфат калия:
NH2SO3H + KHSO4 + Са(ОН)2 = NH2SО3K+CaSО4 + 2Н2О.
Затем раствор калиевой соли сульфаминовой кислоты фильтруют на воронке 6 от CaSО4. Фильтрат передают в вакуумную выпарную колонку 7, где он упаривается и далее поступает в кристаллизатор 8 и на воронку 9. Отжатые кристаллы калиевой соли сульфаминовой кислоты передают в конденсатор 10, куда из мерника 11 приливают формалин. Реакция конденсации производится при 30°С, рН раствора поддерживается равным 5. Затем раствор упаривают в вакуум-выпарном аппарате 12, охлаждают в кристаллизаторе 13 и кристаллы отфильтровывают на воронке 14. Полученный циклотриметилениминосульфонат калия («белую соль») сушат в сушилке 15.
Нитрование «белой соли» ведут нитросмесью состава: 80–81% HNO3; 4–5% Н2SO4; 13–14% SО3 и 1–2% N2O4, подаваемой из мерника 16 (в количестве 1,8 вес. ч. на 1 вес. ч. «белой соли») в нитратор 17. Нитромасса из нитратора 17 передается в разбавитель 18, куда из мерника 19 подается вода для разбавления до удельного веса 1,2–1,3. Затем на воронке 20 гексоген отфильтровывают от отработанной кислоты и после предварительной промывки водой передают на окончательную промывку и стабилизацию. Отработанная кислота имеет состав: 23% HNO3; 13–14% H2SO4; 10–11% KHSO4; 52–54% H2O [21].
Выход гексогена составляет 80% от теоретического, считая на циклотриметилениминосульфонат калия. Этот метод, несмотря на высокий выход гексогена, имеет ряд существенных недостатков, главным из которых является значительная опасность, так как для нитрования используется высококонцентрированная серно-азотная кислотная смесь.
Получение гексогена методом «К»
В 1936 г. Кноффлером был разработан метод производства гексогена, названный методом «К». Принципиальная схема технологического процесса получения гексогена по этому методу изображена на рис. 7 (см. приложение).
Уротропин нитруется раствором аммонийной селитры в концентрированной азотной кислоте. Нитрование осуществляется в две стадии. В первой стадии к раствору аммонийной селитры в азотной кислоте при температуре 20° добавляют уротропин. При этом образуется гексоген и формальдегид. Последний во второй стадии при температуре 65–70°С взаимодействует с аммонийной селитрой и азотной кислотой, образуя добавочное количество гексогена. Далее массу охлаждают.
Полученный гексоген отделяют от отработанной кислоты на барабанном фильтре, промывают водой и кристаллизуют из ацетона. В случае необходимости гексоген подвергают флегматизации.
Отработанная кислота содержит некоторое количество формальдегида, вследствие чего является нестойкой и не может быть подвергнута переработке. Поэтому вначале отработанную кислоту нагревают в специальных аппаратах при 90–95°С. При этом происходит полное окисление формальдегида и частичное разложение аммонийной селитры. Выделяющиеся при этом окислы азота и пары азотной кислоты поступают на абсорбционную установку. Стабилизированную азотную кислоту, содержащую около 48% HNO3 и 24% NH4NO3, подвергают дистилляции в специальных вакуум-аппаратах.
Основным преимуществом метода «К» является хороший выход гексогена (по формальдегиду 60% от теоретического). Серьезными недостатками метода являются: большое количество перерабатываемых материалов (на тонну гексогена перерабатывается свыше 14 т продуктов, что приводит к резкому снижению производительности аппаратуры и усложняет процесс) и весьма сложный процесс регенерации азотной кислоты и аммонийной селитры.
На одну тонну гексогена расходуется: уротропина 0,48–0,5 т, аммонийной селитры 4,8 т; азотной кислоты 8,6 т. Регенерируется аммонийной селитры 3,6 т, азотной кислоты 7,2 т [9, с. 27].
Получение гексогена уксусноангидридным методом
Немецкий вариант
В Германии разработано два метода производства гексогена с применением уксусного ангидрида: метод «КА» и метод «Е».
Метод «КА» явился дальнейшим усовершенствованием метода «К». Технологический процесс получения по методу «КА» складывается из следующих операций: 1) получение динитрата уротропина; 2) получение тринитрата аммония; 3) нитролиз динитрата уротропина тринитратом аммония в присутствии уксусного ангидрида при 50–60°С; 4) фильтрация гексогена от кислот, промывка гексогена и стабилизация его путем варки с водой под давлением; 5) сушка гексогена; 6) регенерация уксусного ангидрида и отработанной кислоты.
Выход гексогена по этому методу по формальдегиду достигает 75 – 80% от теоретического. Недостатками метода является получение гексогена с низкой температурой плавления (не выше 192°С) и опасность регенерации отработанной уксусной кислоты, так как в ней содержатся нестойкие продукты.
На 1 т гексогена расходуется: уротропина 0,4 т, аммонийной селитры 0,43 т, азотной кислоты (99%) 0,68 т и уксусного ангидрида 2,4 т.
Другой метод получения гексогена был разработан в 1935–1938 гг. Эльбе и получил название метода «Е». Исходными продуктами являются параформальдегид, аммонийная селитра, уксусный ангидрид и в качестве катализатора фтористый бор. Процесс осуществляется следующим образом.
В уксусный ангидрид, содержащий фтористый бор (0,4% BF3) и нагретый до 60–65°С, при энергичном перемешивании вводят параформальдегид и сухую аммонийную селитру. Полученный гексоген отделяют от отработанной кислоты на нутч-фильтре. Отработанную уксусную кислоту дистиллируют и переводят снова в уксусный ангидрид, который возвращают в цикл. Преимуществами способа являются большой выход гексогена (80% от теоретического, считая по формальдегиду) и применение в качестве нитрующего средства более дешевой, чем концентрированная азотная кислота, аммонийной селитры. Недостатки его – низкая температура плавления получающегося гексогена вследствие содержания в нем примесей (около 7%), опасность регенерации отработанной кислоты; сложность изготовления параформальдегида и больший расход аммонийной селитры и уксусного ангидрида.
На 1 т гексогена расходуется: параформальдегида 0,63 – 0,635 т, аммонийной селитры 1,8 т, уксусного ангидрида 5 – 5,1 т и фтористого бора 0,019 т. Полученный гексоген содержит до 6% октогена.
Канадский вариант
В Канаде, США и Англии проводилась совместная работа по созданию уксусноангидридного метода получения гексогена. В октябре 1940 г. Росс и Шислер обнаружили, что при реакции формальдегида и нитрата аммония с уксусным ангидридом получается гексоген:
ЗСН2O + 3NH4NO3 + 6 (СН3СО)2O = (CH2NNO2)3 + 12CH3COOH.
Проведение этой реакции не требует применения уротропина и больших количеств азотной кислоты. При новом методе используется уксусный ангидрид, который указанные страны имели возможность производить в большом количестве.
Исследования были начаты в Канаде группой, работавшей под руководством Бахмана из Мичиганского университета, и проверялись на канадских предприятиях. Работа Бахмана показала, что реакция Росса может быть объединена с нитролизной реакцией для обеспечения лучших выходов. Так как исходные продукты реакции Росса – формальдегид и нитрат аммония – образуются как отход нитролизной реакции, то дополнительное добавление в нитромассу нитрата аммония и уксусного ангидрида должно было привести к образованию еще одной молекулы гексогена. Это подтвердилось опытами.
Уравнение комбинированного процесса, предложенного Бахманом, может быть представлено следующим образом:
C6H12N4+4HNO3+2NH4NO3+6 (CH3CO)2O=2 (CH2NNO2)3+12CH3COOH
Технологический процесс получения гексогена по способу Бахмана-Росса состоит в следующем [18, с. 285].
Вначале приготовляют растворы уротропина в ледяной уксусной кислоте и нитрата аммония в 97%-ной азотной кислоте. Растворы нагревают до 40°С, одновременно нагревают и уксусный ангидрид до 60°. Приготовленные растворы медленно сливают в уксусный ангидрид. Слив компонентов производят при 70–75°С. По окончании слива смесь выдерживают 15–20 минут при той же температуре, а затем в нитромассу приливают 640 молей нагретой до 40°С воды.
Регенерацию уксусной кислоты производят добавлением к отработанной кислоте серной кислоты в два приема. Первую порцию приливают в количестве 5–10% от веса отработанной кислоты и затем массу нагревают до 100°С. Вторую порцию приливают в количестве 15–40%. Смесь подвергают дистилляции при 120–140°С для отгонки уксусной кислоты. Рекомендуется предварительно отработанную кислоту обрабатывать NH3, чтобы уменьшить содержание HNO3 до 1%. Предложено полученную нитромассу разбавлять водой при 90–100°С и после отделения гексогена подвергать дистилляции для отгонки уксусной кислоты.
1.4 Октоген: свойства, способы получения
Октоген впервые был описан в 1941 году Д.Ф. Райтом, В.Е. Бахманом и X. Фишером, как примесь к гексогену, получаемому по уксусноангидридному варианту.
В СССР впервые в 1951 г. октоген получен в ЛТИ им. Ленсовета В.С. Шпаком, В.Л. Козловой и Н. И, Галкиной. Они выявили основные закономерности его образования при нитролизе уротропина азотной кислотой в среде уксусной кислоты и ангидрида.
В 1960–1965 годах сотрудниками ЛТИ им. Ленсовета (А.А. Яковлев, Б.В. Гидаспов, Ю.В. Павлов, и др.) и сотрудниками НИИ-6 (Л.-С.Ш. Брозголь, Л.И. Витковская, В.И. Парфенов, С.С. Молчанова) была проведена разработка и отработка технологии получения октогена на пилотных установках [5, с. 138].
В 1966 году была поставлена задача по созданию промышленного производства на заводе им. Я.М. Свердлова. Для отработки технологии в короткие сроки в ДНИХТИ в условиях опытного производства была проведена сравнительная проверка способов получения, рекомендованных ЛТИ и НИИ-6. Более технологичным оказался вариант ЛТИ. При отработке процесса было установлено, что наиболее узкой стадией является фильтрование октогена-сырца от отработанной кислоты и промывных вод и стадия кристаллизации (рекристаллизации). Длительность этих стадий (до 24 часов) объяснялась наличием примесей в продукте и высокой дисперсностью кристаллов. Сотрудниками ДНИХТИ В.Т. Пыховым, Н.И. Работинским, Г.М. Хрисановой и др. был разработан процесс стабилизации качества октогена-сырца путем разрушения примесей и увеличения размеров кристаллов при термировании продукта в разбавленной водой реакционной массе. Внедрение этого процесса позволило снизить длительность стадии фильтрования и кристаллизации до 3–5 часов. Одновременно была разработана технология регенерации отработанной кислоты и ацетона.
Приобретенный опыт и найденные технические решения позволили выдать исходные данные для проектирования промышленного производства октогена на заводе им. Я.М. Свердлова. В период проектирования и строительства промышленного производства текущие потребности в октогене обеспечивались опытным производством института.
Во время строительства первой очереди производства октогена на заводе им. Я.М. Свердлова решался вопрос по созданию второй очереди, причем для повышения технико-экономических показателей была решена задача увеличения выхода октогена на 10%. В 1972 году пущена I очередь, а в 1974 году – II очередь производства октогена.
В связи с возрастающей потребностью в октогене и согласно приказу министра машиностроения в 1974 году ДНИХТИ было поручено провести наращивание мощности I очереди по выпуску этого продукта в 1976–1977 годах. Эта задача была выполнена.
Объем производства октогена, получаемого на созданной установке, не обеспечивал возрастающие потребности военной техники в этом продукте и потребовалось в кратчайшие сроки разработать и внедрить высокопроизводительные технологии получения октогена. Увеличение выпуска октогена в 1974–1982 годах проводилось как совершенствованием уже действующего на заводе им. Я.М. Свердлова периодического способа производства, так и разработкой и созданием более производительного непрерывного процесса.
Свойства октогена
Циклотетраметилентетранитроамин (октоген) интересен как возможная примесь к гексогену (до 10%), полученному уксусноангидридным методом. Поэтому знать свойства, а также условия его образования необходимо и для производства гексогена. Это особенно важно с точки зрения техники безопасности, так как октоген имеет четыре полиморфные формы, из которых только одна β-форма устойчива. Необходимость удаления нестойких и более чувствительных изомеров октогена вызывает дополнительные трудности при очистке гексогена, полученного уксусноангидридным методом [17, с. 20].
Октоген является твердым кристаллическим продуктом белого цвета. При медленной кристаллизации из ацетона октоген получается в виде крупных прозрачных кристаллов ромбической формы. Очищенный перекристаллизацией из ацетона продукт имеет температуру плавления 276–277°С (разлагается при плавлении). Некоторые свойства октогена приведены в табл. 1 (см. приложение).
Октоген не растворим в метиловом и этиловом спиртах, в бензоле, толуоле, ксилоле, серном эфире; плохо растворим в дихлорэтане, анилине, нитробензоле, диоксане. Растворимость в воде при 15–20° около 0,003% и при 100° около 0,02%.
Основные взрывчатые свойства октогена:
Температура вспышки 291°С
Расширение в бомбе Трауцля – 415 мл
Скорость детонации – 9124 м/с
Чувствительность к удару по русской пробе – 84% [26].
Способы получения октогена
Октоген и гексоген получают из одних и тех же компонентов: уротропина, азотной кислоты, нитрата аммония, уксусного ангидрида и уксусной кислоты, но различия в результатах достигают изменением соотношения компонентов, порядком их смешивания и температурными условиями синтеза. Нитролиз уротропина в жестких условиях (повышенная температура, большое количество уксусного ангидрида) приводит к образованию, главным образом, шестичленного циклического полинитрамина-гексогена. Такие условия осуществляются при уксусноангидридном способе, если растворы уротропина в уксусной кислоте и нитрата аммония в азотной кислоте приливают к уксусному ангидриду. В более мягких условиях, когда уксусный ангидрид и раствор нитрата аммония в азотной кислоте приливают к раствору уротропина в уксусной кислоте, образуется восьмичленный циклический полинитрамин-октоген с небольшой примесью гексогена. При синтезе октогена используют примерно втрое меньше количество уксусного ангидрида, чем при синтезе гексогена. [16, с. 16].
Температурные условия и среда, в которой синтезируют октоген, как правило, приводят к образованию β-модификации. Поэтому технологический процесс получения октогена включает стадию рекристаллизации α-модификации в β-модификацию.
Динитропентаметилентетрамин (ДПТ) является промежуточным продуктом нитролиза уротропина до октогена и в значительной степени выход ДПТ определяет выход октогена.
ДПТ может быть получен не только из уротропина, и это расширяет возможности синтеза октогена. Предложено два варианта синтеза октогена: прямой синтез из уротропина в одну стадию; синтез из уротропина в две стадии с выделением продукта первой стадии (ДПТ). Синтез октогена из уротропина в одну стадию
Октоген получают добавлением к раствору уротропина (0,5 моля) в уксусной кислоте при 50–55°С одновременно из двух капельных воронок раствора нитрата аммония (1 моль) в 99% азотной кислоте (2 моля) (так называемый тринитрат аммония) и уксусного ангидрида (3,5 моля), таким образом, чтобы вначале получался незначительный избыток тринитрата аммония. Выход октогена составляет 40%, если из одного моля уротропина должно получиться 1,5 моля октогена:
2C6H12N4 + 8HNO3 + 4NH4NO3+12 (CH3CO)2O → 3C4H8(NNO2)4 + 24СН3СООН
и 60% при условии получения 1 моля октогена из 1 моля уротропина.
Синтез октогена в две стадии с выделением промежуточного продукта ДПТ
На первой стадии процесса синтезируют ДПТ, являющийся по существу промежуточным продуктом и в одностадийном синтезе. ДПТ представляет собой белое кристаллическое вещество, температура плавления 205–206°С с разложением, удельный вес 1,63 г./см. ДПТ плохо растворим в воде и большинстве органических растворителей. Его можно перекристаллизовать из ацетона, этилацетата, нитрометана, диоксана и уксусного ангидрида. В уксусной кислоте ДПТ хорошо растворяется [20].
Первая стадия – получение ДПТ. Впервые ДПТ был выделен путем нейтролизации щелочным ацетоном отработанной кислоты производства гексогена. В условиях реакции Хейла ДПТ был получен по следующей методике: в трехгорловую колбу или алюминиевый реактор объемом 500 см, снабженную мешалкой и термометром, помещают азотную кислоту (133 г. или 2,1 моля концентрации 99,6%) и при 20–25°С (охлаждение водой со льдом) вносят с постоянной скоростью при помощи шнека-питателя уротропин (14,0 г, 0,1 моля) в течение 12–15 мин [8].
При поведении реакции в стеклянном аппарате для безопасности процесса лучше вместо уротропина использовать его динитрат с соответствующим уменьшением количества азотной кислоты. Полученную суспензию выдерживают при перемешивании 5–15 мин, выливают в 1 кг льда и фильтруют. В холодный фильтрат при перемешивании и охлаждении добавляют 15–20% аммиака. При достижении в растворе рН=5,6 медленно образуется осадок ДПТ. При достижении рН=6,5 массу выдерживают 10–15 мин и осадок отфильтровывают. В фильтрат, чтобы обеспечить закономерность осаждения, дополнительно дают аммиак до получения среды рН=7. Выход ДПТ с температурой плавления 177–179°С достигает 12% (2,6 г) из расчета одного моля на моль уротропина.
ДПТ можно получить действуя на нитрат уротропина серной кислотой. Для этого в реактор с 90%-ной серной кислотой добавляют динитрат уротропина при 8–15°С и через 45 мин выдержанную массу выливают на лед и фильтруют. Фильтрат нейтрализуют 28% раствором аммиака до рН=5,6–6,5 и получают ДПТ с выходом 31% от теоретического. Известны методы получения ДПТ из других исходных веществ.
Получение ДПТ из динитрата уротропина и уксусного ангидрида [17, с. 96].
Смесь уксусного ангидрида (20 молей) и динитрата уротропина (5 молей) перемешивается в течении двух-трех дней. Через несколько часов начальное повышение температуры прекращается и начинается ее падение. Осадок отфильтровывают и промывают водой с выделением чистого 224 г. ДПТ, плавящегося при 205–206°С. фильтрат выливают в 10%-ную уксусную кислоту и получают дополнительно 109г ДПТ с температурой плавления 187–200°С. общий выход составляет 30,6% от теоретического.
Уравнение реакции образования ДПТ может быть представлено следующим образом:
C6H12N4 2НNO3 + 3 (СН3СО)2O→ C5H10N6O4 + Н2С(ООССН3)2 + 4СН3СООН
ДПТ получают также путем одновременной и пропорциональной дозировки раствора уротропина (1 моль) в уксусной кислоте (3,1 моля) и раствора азотной кислоты (2 моля) в уксусном ангидриде (2,6 моля) в небольшое количество уксусной кислоты. Время смешивания компонентов 30 минут. Температура – 25–30°С. Выход ДПТ составляет 45–48%.
Аналогичным способом, но с большим количеством уксусной кислоты (23,2 моля) и уксусного ангидрида (4,35 моля) с одновременным добавлением нитрата аммония (1,5 моля), был получен ДПТ смешиванием компонентов при 44°С в течение 15 мин. Эти изменения позволили увеличить выход ДПТ до 65%.
Получение ДПТ из диметилолнитрамида и смеси формальдегид-аммиак
К 37%-ному раствору формалина (0,006 моля), охлажденному до 0°С, добавляют свежеприготовленный нитрамид (0,004 моля). Затем добавляют воду и медленно подливают 10%-ный раствор аммиака до рН=7. Массу выдерживают при 0°С в течение 40 мин. Полученный осадок отфильтровывают, промывают холодной водой и сушат при 50°С. Выход ДПТ 83% по формальдегиду, температура его плавления 206–208°С (с разложением).
Получение ДПТ из диметилолнитрамида и метилендиамина
К холодному раствору, приготовленному смешиванием нитрамида (0,0016 молей) и 37%-ного раствора формалина (0,0016 молей), добавляют сульфат метилендиамина вместе с водой. Полученный раствор охлаждают до 0°С и нейтрализуют до рН=7. Выход ДПТ 97% по формальдегиду.
Вторая стадия – нитролиз ДПТ до октогена. Октоген получен обработкой ДПТ:
1) концентрированной азотной кислотой (99%-ной при 10°С);
2) смесью азотной кислоты с нитратом аммония;
3) смесью концентрированной азотной кислоты с нитратом аммония и уксусным ангидридом.
Второй способ проводится в двух вариантах. Согласно первому варианту в смесь из нитрата аммония и концентрированной азотной кислоты (3 и 7 молей соответственно) добавляют ДПТ (1 моль) при 70°С в течение 15 мин, затем выдерживают 20 мин при 60°С и 10 мин при 25°С, далее смесь выливают на лед. Влажный осадок нагревают с 70%-ной азотной кислотой до прекращения выделения бурых паров, охлаждают и выливают в воду. Выход октогена 65% от теоретического, считая по ДПТ.
Согласно второму варианту в смесь нитрата аммония и концентрированной азотной кислоты (1,6 и 3,2 моля соответственно) добавляют ДПТ (1 моль) при 60–65°С в течение 1 ч, затем добавляют воду и нагревают 12 ч на паровой бане. Выход октогена 75% от теоретического по ДПТ.
По третьему способу к суспензии ДПТ (0,046 моля) в уксусном ангидриде (0,394 моля) при 65–70°С в течение 15 мин подливают раствор нитрата аммония (0,138 моля) в концентрированной азотной кислоте (0,319 моля), выдерживают 20 мин при 65°С и 10 мин при 25°С, затем выливают на лед, отфильтровывают осадок, кипятят с 70%-ной азотной кислотой (203 мл) до прекращения выделения бурых паров. Продукт отфильтровывают, промывают и высушивают. Выход октогена 50% с температурой плавления 267–268°С.
Октоген как термостойкое ВВ используется в зарядах для перфорации глубоких нефтяных скважин, в термостойких капсюлях-детонаторах, детонирующих шнурах. В США его применяют при температуре до 210°С, в основном при прострелочно-взрывных работах, а также при дроблении горячих слитков, разгрузке и ремонте доменных печей и т.п.
2. Методическая разработка факультативных занятий по химии
На основе дипломной работы были разработаны факультативные занятия в виде лекций по теме Бризантные взрывчатые вещества для учащихся старших классов средней общеобразовательной школы.
Задачи факультативных занятий:
1. Повысить познавательный интерес учащихся к химии;
2. Показать учащимся потенциальную опасность взрывчатых веществ;
3. Расширить кругозор учащихся в области взрывчатых веществ.
Занятие 1
Тема занятия: Бризантные взрывчатые вещества. История. Классификация.
Цель занятия: Дать общее понятие о бризантных взрывчатых веществах.
План занятия:
1. Что такое взрывчатые вещества.
2. Возникновение и развитие производства взрывчатых веществ.
3. Классификация взрывчатых веществ.
Ход занятия:
1. Взрывчатыми называют вещества (химические соединения) или их смеси, способные к крайне быстрому самораспространяющемуся химическому превращению с выделением тепла и образованием газообразных продуктов. Взрывчатыми могут быть вещества любого агрегатного состояния – твердые, жидкие, газообразные, в том числе суспензии, эмульсии, коллоидные растворы. Наиболее широко применяются взрывчатые вещества твердого и жидкого агрегатного состояний.
Все процессы, сопровождающиеся образованием в окружающей среде скачка давления (ударной волны), обобщенно называют взрывом.
2. Промышленность взрывчатых веществ возникла во второй половине девятнадцатого века, но значительное развитие она получила лишь в двадцатом столетии на базе бурного роста основной химической промышленности, а также коксохимической и нефтеперерабатывающей отраслей промышленности – источников органического сырья для основных бризантных взрывчатых веществ.
Первыми возникли производства нитратов спиртов – пироксилина и нитроглицерина. В конце XIX и начале XX вв. на вооружение были приняты нитропроизводные ароматических соединений (пикриновая кислота, тротил, тетрил и др.), свойства которых делали значительно менее опасными их производство и обращение с ними. Последнее обстоятельство позволило их широко применять для снаряжения снарядов.
В период подготовки к первой мировой войне страны, развязавшие ее, заготовили большое количество боеприпасов. Однако расход снарядов оказался более значительным, чем это предполагалось. Стало ясно, что именно артиллерийский огонь является самым губительным и наносит наибольшие потери.
Большой расход боеприпасов во время первой мировой войны вызвал рост промышленности взрывчатых веществ. На вооружение были приняты многие полинитросоединения ароматических углеводородов, получающихся при коксовании каменного угля и пиролизе нефти. Чтобы удовлетворить возросшие потребности во взрывчатых веществах, для снаряжения снарядов стали широко применять смеси главным образом на основе аммонийной селитры.
Вторая мировая война потребовала еще большего количества взрывчатых веществ. Расход их значительно превысил расход во время первой мировой войны.
В связи с острым недостатком взрывчатых веществ делались попытки расширить их номенклатуру путем использования новой сырьевой базы, однако эти попытки успеха не имели.
Основным бризантным взрывчатым веществом во время второй мировой войны был тротил. Если в первую мировую войну применялись в значительных количествах и другие нитропроизводные ароматических углеводородов, то во вторую мировую войну тротил или взрывчатые смеси на его основе использовались особенно широко. Так, удельный вес производства тротила в германской промышленности составлял более 50% производства всех взрывчатых веществ.
Широкое применение во время второй мировой войны приобрели наиболее мощные взрывчатые вещества – гексоген и тэн, которые в первой мировой войне не применялись. Особенно большое внимание было уделено развитию промышленности этих веществ в странах, не обеспеченных в достаточной мере собственными источниками сырья для производства взрывчатых веществ на базе ароматических соединений. Например, в Италии гексоген и тэн начали изготовлять в массовом масштабе уже в 1932–1933 гг. оба эти вещества использовались итальянцами для снаряжения боеприпасов не только в чистом виде, но и в смесях с аммонийной селитрой. В Германии широко применяли для снаряжения бронебойных и кумулятивных снарядов. Тэн и гексоген производили также во Франции, Англии, Канаде, США.
3. По составу бризантные взрывчатые вещества разделяются на две большие группы: химические соединения и взрывчатые смеси.
В первую группу входят следующие важнейшие классы взрывчатых веществ:
1) нитросоединения органические соединения, содержащие одну или несколько групп C-NO2 (тротил). Наибольшее значение имеют полинитропроизводные ароматических соединений. Значительная часть нитросоединений алифатических углеводородов также является ВВ, но лишь немногие из них имеют практическое значение;
2) нитроамины органические соединения, содержащие группы N-NO2 (гексоген, тетрил);
3) эфиры азотной кислоты – органические соединения, содержащие группы О-NO2 (тэн) В качестве ВВ применяются азотнокислые эфиры спиртов и углеводов;
4) соли азотной кислоты. Наибольшее применение имеет нитрат аммония (NH4NO3), а также нитраты органических оснований (мочевины, метиламина и др.).
Вторую группу составляют взрывчатые смеси, содержащие и не содержащие ВВ.
К важнейшим классам взрывчатых смесей, содержащим взрывчатые компоненты, относятся:
1) аммониты или аммонийноселитренные ВВ, состоящие из смеси аммонийной селитры с нитросоединениями;
2) сплавы и смеси нитросоединений;
3) нитроглицериновые ВВ (динамиты);
4) хлоратные и перхлоратные ВВ – смеси хлорноватой или хлорной кислот с нитросоединениями и др.
В состав смесей, состоящих из невзрывчатых компонентов, входят горючие вещества и соединения, содержащие значительное количество кислорода или другого окислителя. Реакция взрыва в этом случаи заключается в окислении элементов, входящих в горючие вещества, кислородом, содержащимся в окислителях. Взрывчатые смеси из невзрывчатых компонентов могут быть разбиты на следующие классы:
1) дымные пороха – смеси селитры и угля;
2) оксиликвиты смеси жидкого кислорода с горючими веществами;
3) смеси концентрированной азотной кислоты или другого жидкого окислителя с горючими веществами.
Из взрывчатых смесей наибольшее значение имеют аммонийноселитренные ВВ.
Артиллерия предъявляет очень жесткие требования к бризантным ВВ. Они должны обладать большой мощностью, быть безопасными в обращении, иметь достаточную чувствительность к начальному импульсу, быть стойкими при хранении. Кроме всего перечисленного, бризантное ВВ, принятое на вооружение, должно быть обеспечено сырьевой базой и метод производства его должен быть достаточно прост и безопасен.
Применяемые в настоящее время взрывчатые вещества далеко не в полной мере удовлетворяют перечисленным требованиям и поэтому изыскание новых мощных взрывчатых веществ, обладающих указанными выше свойствами, является важной задачей ученых и инженеров, работающих в этой области. Одновременно актуальна и проблема усовершенствования технологии производства ВВ с целью снижения опасности их изготовления и повышения производительности труда, и как следствие – снижение себестоимости продукта.
Занятие 2
Тема занятия: Тротил. Тэн.
Цель занятия: Дать учащимся общее представление о тротиле, о тэне, познакомить со свойствами и методами получения тротила, тэна.
План занятия а:
1. Свойства тротила и тэна.
2. Получение тротила и тэна.
3. Применение тротила и тэна.
Ход занятия:
1. Тротил по внешнему виду представляет собой желтое вещество. Температура плавления очищенного тротила 80,6°С, при наличии примесей температура плавления снижается до 75–77°С. Примеси образуют с тротилом многокомпонентные эвтектические сплавы, имеющие маслообразный вид, вследствие чего их называют тротиловым маслом.
Плотность монокристалла тротила 1,663 г./см. Гигроскопичность около 0,05%, растворимость в воде низкая – 0,15% при 100°С, что является благоприятным свойством.
Тротил токсичен, предельно допустимая концентрация 0,001 мг/л, он поражает дыхательные пути, пищеварительный тракт. При длительном воздействии вызывает слабость, головокружение, дерматиты кожи, гепатит,
Тэн представляет собой белое кристаллическое вещество с температурой плавления 141–142°С и плотностью 1,77 г./см, плохо прессуется. Прессованием можно достичь плотности 1,6 г/см. Размер частиц 10–830 мкм.
Тэн не гигроскопичен, растворимость его в воде при 19°С 0,01%, а при 100°С – 0,035%. Тэн химически стоек, более чувствителен к механическим воздействиям, чем гексоген и октоген. Температура вспышки 205–225°С.
Тэн – токсическое вещество, вызывает раздражение верхних дыхательных путей, покраснение слизистых оболочек и кожи; при попадании в легкие вызывает расширение кровеносных сосудов.
2. Тротил получают нитрацией толуола смесью азотной и серной кислот.
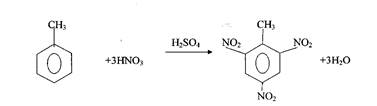
Тэн – сложный эфир азотной кислоты и четырехатомного спирта – пентаэритрита С(СН2ОН)4.
С(СН2ОН)4 + 4HNО3 → С(СН2ONО2)4 + 4Н2О
3. Тротил в чистом виде или смеси с гексогеном (смеси ТГ, состав В) или тэном (пентолит) широко применяется в виде литых и прессованных шашек в качестве промежуточных детонаторов, кумулятивных зарядов для дробления негабаритных кусков породы, зарядов для сейсморазведки.
Чистый тэн используется для снаряжение средств инициирования и детонирующих шнуров, для прессованных дополнительных детонаторов, а также в эластичных ВВ.
Занятие 3
Тема занятия: Гексоген. Октоген.
Цель занятия: Дать учащимся общее представление о гексогене и октогене, познакомить со свойствами и методами получения гексогена и октогена.
План занятия:
1. Свойства гексогена и октогена.
2. Получение гексогена и октогена.
3. Применение гексогена и октогена.
Ход занятия:
1. Гексоген по внешнему виду представляет собой белое вещество с плотностью монокристалла 1,816 г/см3. При прессовании достигает плотность 1,73 г/см3. Температура плавления 204,5–205°С. Гексоген практически не гигроскопичен. Он весьма ядовит, предельно допустимая концентрация 0,001 мг/л; поражает центральную нервную систему, главным образом головной мозг, вызывает нарушения кровообращения и малокровие.
Гексоген характеризуется высокой чувствительностью к механическим воздействиям. С целью снижения чувствительности гексоген флегматизируют воскоподобными веществами.
Температура вспышки 220–230°С. На открытом воздухе он горит ярким белым пламенем без остатка, при быстром нагревании разлагается со взрывом.
Температуру 185°С выдерживает в течение 2,5 ч.
Октоген впервые был обнаружен как примесь к гексогену. Долгое время он интересовал исследователей исключительно как вещество, сопровождающее гексоген. Однако в последние годы его начали изучать как самостоятельное ВВ, так как октоген, имея все положительные качества гексогена, выгодно отличается от него более высокой термостойкостью, большей плотностью и соответственно лучшими взрывчатыми характеристиками.
Октоген – белое кристаллическое высокоплавкое вещество с плотностью монокристалла 1,906 г/см3. Температура плавления 278,5–280°С. Октоген не гигроскопичен, в воде при 15–20°С растворяется около 0,003%.
По токсичности октоген аналогичен гексогену.
Октоген, как и гексоген характеризуется высокой чувствительностью к механическим воздействиям.
Температура вспышки октогена 291°С. Октоген отличается сравнительно высокой термостойкостью: температуру 200°С выдерживает 8 ч 30 мин, 205°С – 4 ч 30 мин, 220°С - 2 ч.
2. Гексоген получают нитрацией уротропина азотной кислотой. В общем виде реакцию можно записать следующим образом:
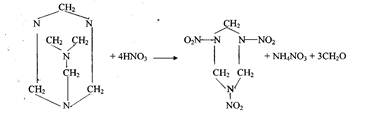
Однако малый выход гексогена и большой расход HNO3 свидетельствует о том, что эта реакция значительно сложнее и сопровождается побочными продуктами.
Октоген получают нитролизом уротропина азотной кислотой в среде уксусного ангидрида и нитрата аммония.
2C6H12N4 + 8HNO3 + 4NH4NO3 + 12 (СН3СО)2O →3C4H8(NNО2)4 + 24CH3COOH
3. Гексоген применяется для снаряжения зарядов малого калибра, кумулятивных зарядов, в детонаторах, в капсюлях-детонаторах. В смеси с алюминиевой пудрой или с тротилом его используют для снаряжения различных боеприпасов.
Гексоген используют также в так называемых пластичных ВВ или во взрывчатых замазках. Смеси из гексогена и связывающего материала являются мягкими, пластичными и немного клейкими. Они применяются для подрывных целей, например, с их помощью можно резать металл, мостовые фермы, ткани и т.п.
Октоген как термостойкое ВВ используется в зарядах для перфорации глубоких нефтяных скважин, в термостойких капсюлях-детонаторах, детонирующих шнурах. В США его применяют при температуре до 210°С, в основном при прострелочно-взрывных работах, а также при дроблении горячих слитков, разгрузке и ремонте доменных печей и т.п.
Заключение
На основе дипломной работы можно сделать следующие выводы:
1. Изучен широкий ряд литературных источников о технологиях штатных взрывчатых веществ и о их свойствах.
2. На основе полученного материала разработаны факультативные занятия для учеников старших классов средней школы.
3. Использование данного материала при проведении факультативных занятий позволило расширить знания учащихся, совершенствовать их научное мировоззрение, выработать у школьников представление о химических технологиях взрывчатых веществ и опасности, при обращении с ними.
Литература
1. Андреев К.К. Взрыв и взрывчатые вещества. – М.: Недра, 1956. – 119 с.
2. Андреев К.К. Термическое разложение и горение взрывчатых веществ. – М.: Наука, 1966. – 346 с.
3. Андреев К.К., Горбунов В.В. Об устойчивости нормального горения порошкообразных взрывчатых веществ. – В сб.: «теория взрывчатых веществ. М.: Высшая школа, 1967. – С. 135–149.
4. Бенсон С. Основы химической кинетике. - М.: Мир, 1964. – 350 с.
5. Взрывчатые вещества, пиротехника, средства инициирования в послевоенный период. Научное издание. Издательство «Гуманистика», М-СПб, 2001.
6. Горст А.И. Химия и технология нитросоединений. – М.: Оборонгиз, 1940. – С. 341.
7. Дубнов Л.В., Бахаревич Н.С, Романов А.И. Промышленные взрывчатые вещества. - М.: Недра, 1973. 358 с.
8. Збарский В.Л., Максимов Ю.Я., Орлова Е.Ю. О влиянии способа очисткина термическую стойкость октогена в сб. «Теория взрывчатых веществ», – М.: Высшая школа, 1967. – С. 84–92.
9. Зуйков А.И., Герасимов В.А. Промышленные взрывчатые вещества и средства взрывания. – Тула.: Мир, 1979. – 63 с.
10. Коротксявич К.Н. Взрывное дело в мирном применении. – М.: Оборонгиз, 1927.
11. Кук, Мелвин А. Наука о промышленных взрывчатых веществах. – М.: Недра, 1980. – 455 с.
12. Максимов Ю.Я. Термический распад нитропроизводных бензола. – в кн.: Теория взрывчатых веществ. – М.: Оборонгиз, 1963. – С. 338–340.
13. Мержанов А.Г., Абрамов В.Г. Тепловой взрыв взрывчатых веществ и порохов. - Черноголовка, 1979.
14. Орлова Е.Ю. Исследование кинетики нитрования в гетерогенных условиях. Сборник докладов на VIII Менделеевском съезде. Секция органической химии. - М: изд. АНСССР, 1959, 2. С. 236.
15. Орлова Е.Ю. Нитрация динитротолуола в гетеролгенных условиях. Тезисы докладов на конфер. МХТИ. М.: изд. МХТИ, 1945.
16. Орлова Е.Ю., Орлова Н.А., Жилин В.Ф. Октоген, получение, свойства и применение. – М: изд. МХТИ им. Д.И. Менделеева, 1970. – 57 с.
17. Орлова Е.Ю., Орлова Н.А., Жилин В.Ф. Октоген термостойкое взрывчатое вещество. – М.: Недра, 1975.
18. Орлова Е.Ю. Химия и технология бризантных взрывчатых веществ.-М.: Оборонгиз, I960. – 396 с.
19. Росси Б.Д., Поздняков 3.Г. Промышленные взрывчатые вещества и средства взрывания. Справочник. – М.: Недра, 1971. 176 с.
20. Светлов Б.Я., Яременко Н.Е. Теория и свойства промышленных взрывчатых веществ. – М.: Недра, 1973.
21. Слипко К.К., Будлинов М.А. Взрывчатые вещества. - М.: Оборонгиз, 1939.
22. Снитко К.К. Взрывчатые вещества, краткий курс. - М.: Недра, 1939.
23. Соколов Н.А. Курс теории взрывчатых веществ. – М.: ОНТИ, 1937. – 384 с.
24. Тамбиев Г.И., Бейсбаев А.М. Технология приготовления и применения простейших ВВ. – М.: Недра, 1996.
25. Ушаков М. Справочник для инженеров и мастеров по производству взрывчатых веществ. – М.: Госхимтехиздат, 1934. С. 171–184.
26. Холево Н.А. Чувствительность взрывчатых веществ к удару. – М.: Мир, 1974. – 136 с
Приложения
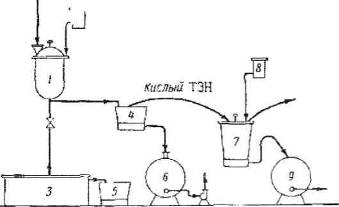
Рис. 1. Схема получения тэна одностадийным способом: 1-нитратор, 2-мерник азотной кислоты, 3-аварийный чан, 4 и 5 – вакуум-воронки, 6-вакуум – сборник отработанной кислоты, 7-разбавитель, 8-бак для воды, 9-вакуум – сборник промывной воды
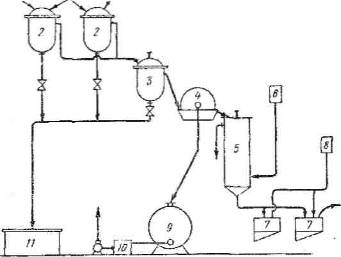
Рис. 2. Схема получения тэна с непрерывным отжимом отработанной кислоты: 1-напорный бак азотной кислоты, 2-основные нитраторы, 3 – буферный нитратор, 4-вакуум-фильтр, 5-промывная колонна, 6 и 8 – напорные баки для воды, 7-вакуум-воронка, 9-отстойник отработанной кислоты, 10-сборник, 11-аварийный бак
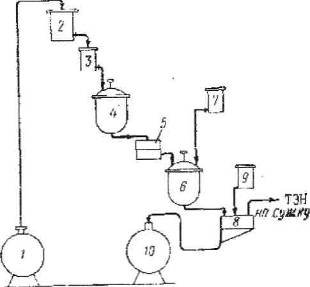
Рис. 3, Схема перекристаллизация тэна: 1-хранилище ацетона, 2-мерник-автомат ацетона, 3-подогреватель, 4-растворитель, 5-фильтр, 6-кристаллизатор, 7 и 9-мерник воды, 8 – вакуум-воронка, 10-сборник маточного ацетона
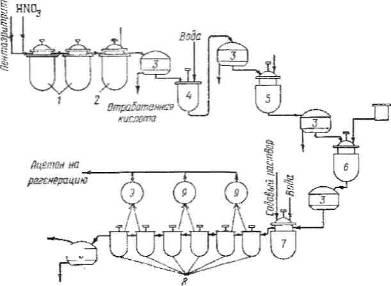
Рис. 4 Схема получения тэна с периодическим отжимом отработанной кислоты: 1-нитраторы, 2-разбавитель, 3-фильтры, 4-промывной аппарат, 5 – нейтрализатор, 6-растворитель, 7-кристаллизатор, 8-дистилляторы, 9 – конденсаторы ацетона
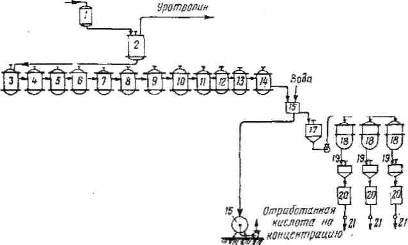
Рис. 5. Схема получения гексогена окислительным методом: 1-бак для охлаждения HNО3, 2-нитратор, 3 по 8-буферные аппараты для донитоовывания, 9, 10 – аппараты для окисления, 11 по 14-охладники, 15 – центрифуга, 16-приемник, 17 – метатель, 18-автоклавы, 19-аппарат для флегматизации, 20-фильтр-воронки, 21-мешки
Рис. 6. Схема получения гексогена через сульфаминовую кислоту: 1 и 10-конденсаторы, 2 и 4-чаны для разложения, 3, 6, 9, 14 и 20 – фильтр-воронки, 7 и 12-выпарные колонны, 8 и 13-кристаллизаторы, 5, 11, 16 и 19 – мерники, 15-сушилка, 17-нитратор, 18-разбавитель, 19 – приемник
© 2009 База Рефератов