
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Курсовая работа: Синтез и свойства адипиновой кислоты
Курсовая работа: Синтез и свойства адипиновой кислоты
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ АДИПИНОВОЙ КИСЛОТЫ”
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
1. Введение
1.1. Свойства адипиновой кислоты
1.2. Применение адипиновой кислоты
1.3. Синтез адипиновой кислоты
2. Литературный обзор. Методы получения дикарбоновых и поликарбоновых кислот
2.1. Карбоксилирование и алкоксикарбонилирование
2.2. Реакции конденсации
2.3. Реакции Михаэля
2.4. Окислительные методы
3. Методика эксперимента
4. Выводы
Список литературы
1. Введение
1.1. Свойства адипиновой кислоты
Адипиновая кислота (1,4-бутандикарбоновая кислота) НООС(СН2)4СООН,
молекулярная масса 146,14; бесцветные кристаллы; т. пл. 153°С, т. кип.
265°С/100 мм рт. ст.; легко возгоняется; d418 =1,344; т.
разложения 210-240°С; ![]() (
(![]() ) = 4,54 (160°С), 2,64 (193
°С);
) = 4,54 (160°С), 2,64 (193
°С); ![]() ;
; ![]() ,
, ![]() ,
, ![]() . Растворимость в воде (г
на 100 г): 1,44 (15°С), 5,12 (40°С), 34,1 (70°С). Растворимость в этаноле, в
эфире – ограниченно.
. Растворимость в воде (г
на 100 г): 1,44 (15°С), 5,12 (40°С), 34,1 (70°С). Растворимость в этаноле, в
эфире – ограниченно.
Адипиновая кислота обладает всеми химическими свойствами, характерными для карбоновых кислот. Образует соли, большинство из которых растворимы в воде. Легко этерифицируется в моно- и диэфиры. С гликолями образует полиэфиры. Соли и эфиры адипиновой кислоты называются адипинатами. При взаимодействии с NH3 и аминами адипиновая кислота дает аммонийные соли, которые при дегидратации превращаются в адипамиды. С диаминами адипиновая кислота образует полиамиды, с NH3 в присутствии катализатора при 300-400 °С – адиподинитрил.
При нагревании адипиновой кислоты с уксусным ангидридом образуется линейный полиангидрид НО[—СО(СН2)4СОО—]nН, при перегонке которого при 210°С получается нестойкий циклический ангидрид (формула I), переходящий при 100°С опять в полимер. Выше 225 °С адипиновая кислота циклизуется в циклопентанон (II), который легче получается пиролизом адипината кальция.

В промышленности адипиновую кислоту получают главным образом двухстадийным окислением циклогексана. На первой стадии (жидкофазное окисление воздухом при 142-145°С и 0,7 МПа) получают смесь циклогексанона и циклогексанола, разделяемую ректификацией. Циклогексанон используют для производства капролактама. Циклогексанол окисляют 40-60%-ной HNO3 при 55°С (катализатор NH4VO3); выход адипиновой кислоты 95%.
Адипиновую кислоту можно получить также:
а) окислением циклогексана 50-70%-ной HNO3 при 100-200°С и 0,2-1,96 МПа или N2O4 при 50°С;
б) окислением циклогексена озоном или HNO3;
в) из ТГФ по схеме:
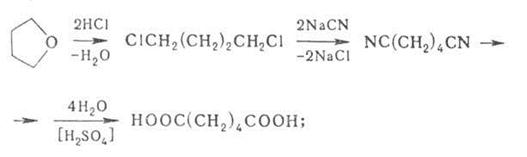
г) карбонилированием ТГФ в ангидрид адипиновой кислоты, из которого действием Н2О получают кислоту.
1.2. Применение адипиновой кислоты
Основная область применения адипиновой кислоты – производство полиамидных смол и полиамидных волокон, а эти рынки давно сформировались и испытывают жесткую конкуренцию со стороны полиэфира и полипропилена [1].
Увеличивается использование адипиновой кислоты в производстве полиуретанов. Сейчас темпы роста производства и потребления полиуретанов превышают темпы роста производства и потребления полиамидов, особенно полиамидных волокон. К примеру, спрос на адипиновую кислоту со стороны западноевропейских продуцентов полиуретана постоянно повышается, и сегодня темпы его роста составляют примерно 12-15 % в год. Тем не менее, спрос на полиамид (нейлон) для производства пластмасс тоже возрастает, особенно в азиатском регионе. Объясняется это тем, что для производства полиуретанов в странах АТР чаще используют простые полиэфиры, в синтезе которых не принимает участия адипиновая кислота, поэтому до 85 % адипиновой кислоты здесь используется в производстве полиамидов. Эта особенность оказывает волновой эффект на спрос адипиновой кислоты в регионе, поэтому среднегодовые темпы прироста мирового спроса на этот продукт прогнозируются на уровне 3-3,5%. В России собственное производство адипиновой кислоты пока отсутствует, хотя имеются весьма благоприятные для этого условия: развита сырьевая база (циклогексанол, циклогексанон, азотная кислота), имеются крупные потребители конечной продукции (пластификаторов, мономеров). Перспективная потребность в адипиновой кислоте для России оценивается величиной в несколько десятков тысяч тонн в год. В Российской Федерации адипиновая кислота используется для производства пластификаторов, полиамидов, фармацевтических препаратов, полиуретанов.
Итак, адипиновая кислота – стратегически и экономически важное сырье в производстве полигексаметиленадипинамида (~ 90% производимой кислоты), ее эфиров, полиуретанов; пищевая добавка (придает кислый вкус, в частности в производстве безалкогольных напитков). То есть продукты на основе адипиновой кислоты находят широкое применение в производство полиамидов, пластификаторов, полиэфиров, полиэфирных смол для ПУ, ППУ, в промышленной переработке стекла, в радиоэлектронной и электротехнической промышленности, в производстве дезинфицирующих средств, в пищевой и химико-фармацевтической промышленности, в получении лаков и эмалей, растворителей, самоотверждающихся составов.
1.3. Синтез адипиновой кислоты
В 5-литровую круглодонную колбу, снабженную механической мешалкой, термометром и делительной воронкой емк. В 1л, помещают 2100г (16,6мол) 50%-ной азотной кислоты (удельный вес 1,32; в вытяжном шкафу). Кислоту нагревают почти до кипения и добавляют 1г ванадата аммония. Пускают в ход мешалку и медленно через делительную воронку добавляют 500г (5мол) циклогексанола. Сперва добавляют 40-50 капель циклогексанола и реакционную смесь размешивают до начала реакции (4-5 мин), что становится заметным по выделению окислов азота (прим. 3). Затем реакционную колбу помещают в баню со льдом, содержимое колбы охлаждают до тех пор, пока температура смеси не достигнет 55-600С. После этого как можно скорее прибавляют циклогексанол, поддерживая температуру в пределах, указанных выше. К концу окисления (после того, как прибавлено 475г циклогексанола) ледяную баню удаляют; иногда колбу приходится даже нагревать для того, чтобы поддерживать необходимую температуру и чтобы избежать циклизации адипиновой кислоты.
Перемешивание продолжают еще час после прибавления всего количества циклогексанола. Затем смесь охлаждают до 0, адипиновую кислоту фильтруют с отсасыванием, промывают 500мл ледяной воды и сушат на воздухе в течение ночи. Выход белых кристаллов с т.пл. 146-1490 составляет 395-410г. Выпариванием маточных растворов можно получить еще 30-40г продукта с т.пл. 141-1440С (примечание 4). Общий выход сырой адипиновой кислоты: 415-440г, или 58-60% теоретич. (прим. 6). Полученный продукт для большинства целей достаточно чист; однако более чистый продукт может быть получен перекристаллизацией сырой адипиновой кислоты из 700мл концентрированной азотной кислоты уд. веса 1,42. потери при очистке составляют около 5%. Перекристаллизованная адипиновая кислота плавится при 151-1520 (примечания 6 и 7).
Примечания.
1. Имеется предположение не применять катализатора, если температуру реакционной смеси, после начала реакции, поддерживать при 85-900 (Хартман, частное сообщение).
2. Применялся технический циклогексанол, практически не содержащий фенола. Более 90% продукта кипело в пределах 158-1630.
3. Весьма важно, чтобы окисление началось до того, как будет прибавлено значительное количество циклогексанола, в противном случае реакция может стать бурной. Необходимо ваести реакцию в хорошо действующем вытяжном шкафу.
4. Азотнокислые маточные растворы содержат значительные количества адипиновой кислоты в смеси с глутаровой и янтарной кислотами. Оказалось, что разделение этих кислот кристаллизацией практически нецелесообразно. Однако, если азотную кислоту удалить выпариванием, а оставшуюся смесь кислот этерифицировать этиловым спиртом,то можно получить смесь этиловых эфиров янтарной (т. кип. 121-1260/20мм), глутаровой (т. кип. 133-1380/20мм) и адипиновой т. кип. (142-1470/20мм) кислоты. Эти сложные эфиры можно успешно разделить перегонкой.
5. Следующая видоизмененная пропись может дать лучший выход. В 3-хлитровую колбу, снабженную мешалкой, обратным холодильником и капелоьной воронкой, укрепленными в асбестовых пробках, пропитанных жидким стеклом, помещают 1900мл 50%-ной азотной кислоты (1262мл азотной кислоты уд. веса 1,42, разбавленной до 1900мл) и 1г ванадата аммония. Колбу помещают на водяную баню, нагретую до 50-600, и очень медленно, при работающей мешалке, прибавляют 357г (3,5мол.) технического циклогексанола таким образом, чтобы температура бани поддерживалась при 50-600. Эта операция продолжается 6-8ч. Реакцию завершают нагреванием водяной бани до кипения, пока не прекратится выделение окислов азота (около 1 часа). Горячую реакционную смесь сливают с помощью сифона и дают ей охладиться. Выход сырой адипиновой кислоты: 372г (72% теоретич.).
Асбестовые пробки, пропитанные жидким стеклом, приготовляют из тонкого асбестового листа, нарезанного в полоски шириной 2,5см. Полоски смачивают раствором жидкого стекла и затем наматывают, например, на форштосс холодильника до получения пробки нужного размера. После сборки прибора пробки покрывают жидким стеклом и оставляют для затвердевания на ночь.
6. Азотнокислые маточные растворы после кристаллизации могут заменять часть свежей кислоты в последующих операциях окисления.
7. Адипиновую кислоту можно также перекристаллизовать из 2,5-кратного (по весу) количества воды или 50%-ного спирта. Однако эти растворители дают менее удовлетворительные результаты, чем азотная кислота.
Другие методы получения.
Адипиновая кислота может быть также получена окислением циклогексана и циклогексанона азотной кислотой или перманганатом калия. Описанный метод основан на патентах Deutsche Hydrierwerke A.-G.
Другие методы получения состоят в окислении циклогексена бихроматом калия и серной кислотой и во взаимодействии γ-броммасляного эфира с натрий-малоновым эфиром с последующим омылением и декарбоксилированием полученного триэтилового эфира 1,4,4-бутантрикарбоновой кислоты.
2. Литературный обзор. Методы получения дикарбоновых и поликарбоновых кислот
2.1. Карбоксилирование и алкоксикарбонилирование
Карбоксильная группа может быть введена двумя путями. Первый путь состоит в применении моноксида углерода в присутствии катализатора, чаще всего металлорганического соединения. Второй путь использует реакцию карбаниона с диоксидом углерода. Оба эти метода мы рассмотрим раздельно.
(1) Карбоксилирование моноксидом углерода
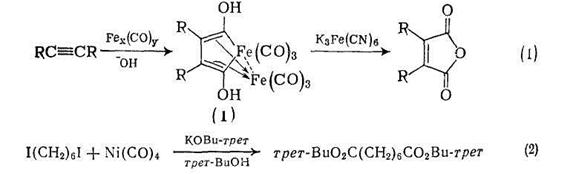
Этому важному методу получения дикарбоновых кислот посвящен обзор [1]. Типичный пример — синтез малеиновых ангидридов при реакции ацетилена с карбонилом железа в водной щелочи {схема (1)}. Продукт реакции (1) при окислении феррицианидом калия или азотной кислотой дает малеиновый ангидрид. Алкоксикарбонилирование органических галогенидов (RHal) карбонилом никеля и алкоксидом щелочного металла разработано Кори [2] и другими авторами, и применяется для синтеза сложных эфиров дикарбоновых кислот {схема (2)}.
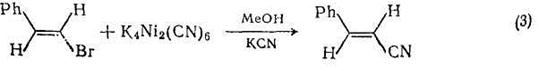
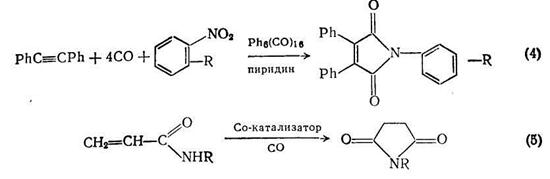
Модификацией этого метода получают мононитрилы {схема (3)}. По-видимому, не существует ограничений для использования этой реакции для синтеза дннитрилов, хотя в оригинальной работе таких примеров не представлено. Малеинимиды можно получать с высоким выходом [3] по реакции дифенилацетилена, моноксида углерода и ароматического нитросоединения с использованием гексадекакарбонилгексародия {Rh6(CO)i6} в качестзе катализатора и третичным амином (пиридин, N-метилпирролидин) в качестве растворителя {схема (4)}. Моноксид углерода, по-видимому, выступает в этих реакциях как восстанавливающий и как карбонилирующий агент; механизм реакций сложен.
Алифатические α,β- и β,γ-непредельные амиды кислот взаимодействуют [4] с моноксидом углерода в присутствии подходящего кобальтового катализатора с образованием имидов янтарной или глутаровой кислот. Лучшим катализатором здесь служит Со2(СО)8, хотя и кобальт Ренея, и ацетат кобальта(II) также катализируют эту реакцию. N-Замещенные акриламиды. с высоким выходом дают соответствующие сукцинимиды {схема (5)}. Аналогично, можно использовать и другие производные акриламида.
(2) Карбоксилирование диоксидом углерода
Превращение металлорганических соединений в соли карбоновых кислот при взаимодействии с диоксидом углерода — хорошо известная реакция [5], с помощью которой {схема (6)} можно проводить как моно-, так и дикарбоксилирование. Образование дикарбоновой кислоты зависит от направления реакции первоначально образующейся натриевой соли фенилуксусной кислоты с локальным избытком бензилнатрия, что приводит к динатриевому производному фенилуксусной кислоты.
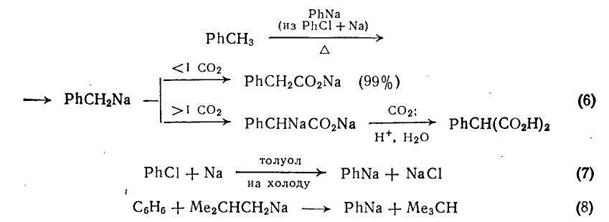
Получению натрий- и калийорганических соединений посвящен обзор [6], где описаны и детали типичных экспериментальных методик. Эти металлорганические соединения можно получать или прямой реакцией доступных органических соединений (обычно галогенида) со щелочным металлом, или реакцией трансметаллирования, которая в основном является кислотно-основной реакцией, оба метода показаны на примере получения фенилнатрия {схемы (7) и (8)}.
Реакции металлирования, включающие литийорганические соединения рассмотрены также в обзоре [7]. Для получения дикарбоновых кислот необходимо использовать бисметаллорганические соединения или металлорганические реагенты, уже содержащие карбоксильную группу. Несмотря на возможность побочных реакций эти превращения применимы к разнообразным соединениям. Далее мы рассмотрим наиболее важные примеры этой реакции.
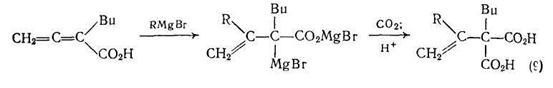
При обработке реактивами Гриньяра некоторые алленкарбоновые кислоты можно превратить в металлорганические соединения. Последующее взаимодействие этих соединений с диоксидом углерода {схема (9)} приводит с хорошим выходом к (1-алкилвинил) малоновым кислотам [8].
Алкилмалоновые кислоты с хорошим выходом {схема (10)} получают при реакции алюминийлитиевого производного карбоновой кислоты (2) с диоксидом углерода [9]; в свою очередь, металлорганпческое производное (2), используемое в этой реакции, получают гидроалюминированием алкинов-1. Например, гексин-1 при взаимодействии с 2 моль диизобутилалюминийгидрида приводит (с 85%-ным выходом) к металлорганическому производному (3) {схема (11)}, которое после обработки метиллитием дает (4). Это соединение реагирует с диоксидом углерода с образованием малоновой кислоты, причем, как показано на схеме (10), реакция идет через образование интермедиата (2).

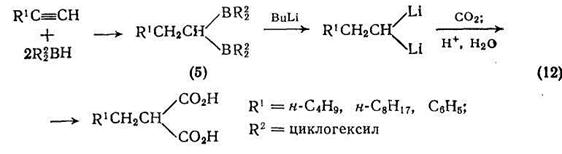
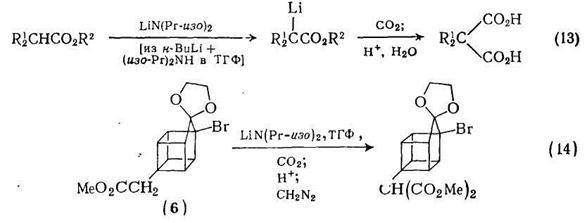
Аналогично, можно проводить превращение ацетиленов в малоновые кислоты с использованием гем-борорганических соединений [10] типа (5) {схема (12)}; при использовании 2 моль бутил-лития можно достичь выхода 65—70%. Другой хороший метод [11] синтеза производных замещенной малоновой кислоты реакция α-анионов сложных эфиров с диоксидом углерода. Анионы генерируют с помощью диизопропиламидалития в тетрагидрофуране,
и дальнейшая процедура сводится к пропусканию диоксида углерода в раствор аниона. Последующая обработка приводит к практически чистому продукту {схема (13)}. Прекрасные результаты получены с такими стерически затрудненными сложными эфирами, как этил-2-метилпропионат; в этом случае побочные реакции не наблюдались. Хорошим примером этой реакции служит синтез адамантан-2,2-дикарбоновой кислоты. Метод можно также использовать в гомокубановой серии; сложный эфир (6) можно превратить в соответствующее производное малоновой кислоты {схема (14)} без деградации или перегруппировки «клеточного» каркаса.
Используя путь, показанный на схеме (15), из бутадиена можно получить набор дикарбоновых кислот. При действии натрия в строго определенных условиях бутадиен димеризуется с образованием динатрийоктадиена. Получающийся делокализованный дианион реагирует с диоксидом углерода, давая смесь трех возможных региоизомерных диеновых дикарбоновых кислот, гидрирование которых приводит к себациновой, 2-этилпробковой и 2,5-диэтиладипиновой кислотам в соотношении 3,5: 5 : 1 соответственно. Эта важная реакция, распространенная на такие ароматические соединения, как стирол и 2-метилстирол, приводит к производным адипиновой кислоты {схема (16)}, причем оба продукта можно гидрировать до соответствующих дициклогексильных производных.
Дианион циклооктатетраена реагирует с диоксидом углерода с образованием дикарбоновой кислоты, однако ранее предложенная для этого продукта структура (7) неверна. Альтернативная формула (8) согласуется с результатами по электроциклическому раскрытию кольца предшественника, имеющего транс-стереохимию, в соответствии с правилом Вудворда — Гофмана о сохранении орбитальной симметрии {схема (17)}.
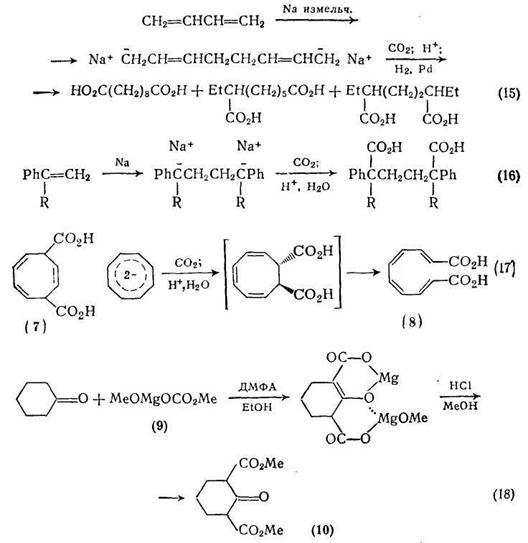
Эффективным реагентом для введения карбоксильной или алкоксикарбонильной группы в различные карбанионы является метилметоксимагний карбонат (ММК) (9). Обычно кетоны превращаются в сложные эфиры а-кетокислот, однако применение избытка ММК может привести к включению двух метоксикарбонильных групп, как, например, при получении синтетически важного диэфира (10) {схема (18)}.
2.2. Реакции конденсации
Большинство общих подходов к синтезу ди- и поликарбоновых кислот использует реакции конденсации. Эти реакции включают сложноэфирную конденсацию Кляйзена и различные реакции производных малоновой и щавелевой кислот.
Производные дикарбоновых кислот с длинной цепью получают из доступных производных дикарбоновых кислот в результате сложноэфирной конденсации Кляйзена. Можно использовать, например, N,N-диметилсебацамат (11) {схема (19)}, так как в конденсацию вовлекаются только сложноэфирная и соседняя с ней α-метиленовая группы.
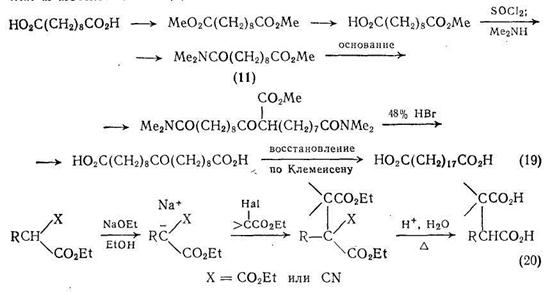
Алкилирование анионов, получаемых из эфиров малоновой кислоты или этилцианоацетата, широко используется для синтеза монокарбоновых кислот, и как видно из схемы (20), может также применяться для получения дикарбоновых кислот. При использовании в качестве алкилирующих агентов соответствующих сложных эфиров галогенокислот {схема (20)} этот метод в принципе может позволить получать различные ди- и поликарбоновые кислоты.
Другое применение диэтилмалоната более специфично, так как реакция диэтилнатриймалоната с соответствующим образом защищенными этилглицидатами приводит к α,β-диэтоксикарбонилбутиролактонам, которые при последующем гидролизе превращаются в параконовые кислоты (12) {схема (21)}. Обработка параконовых кислот полифосфорной кислотой дает соответствующие циклолентен-2-оны-1, включая дигидрожасмон,
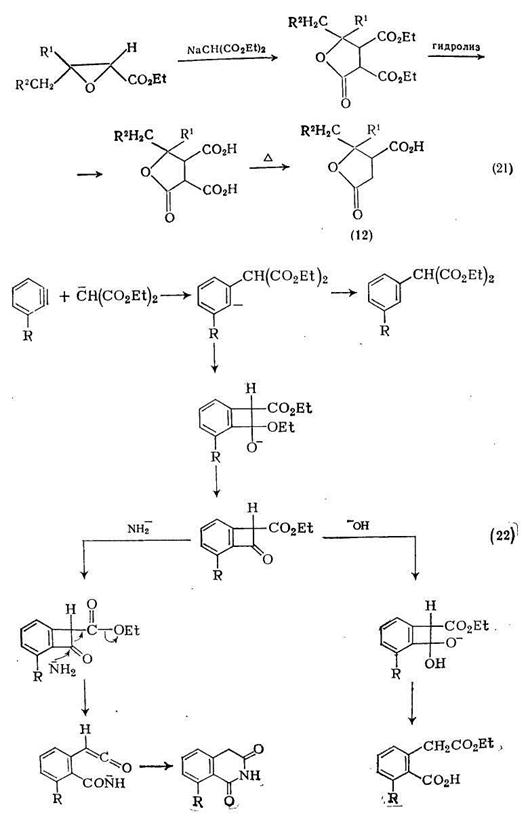
Дегидробензолы реагируют с малоновыми эфирами, давая производные гомофталевой кислоты. Например, реакция диэтилмалоната с о-броманизолом в тетрагидрофуране в присутствии амида натрия с выходом 60% дает 3-метоксигомофталимид; при изменении условий реакции могут появляться другие продукты. При использовании в качестве источника дегидробензола бромбензола и в качестве растворителя гексаметанола основными продуктами реакции являются диэтилфенилмалонат (20%), моноэтилгомофталат (10%) и гомофталимид (50%). Механизм образования этих продуктов показан на схеме (22).
Для синтеза замещенных малоновых эфиров можно использовать прямое алкилирование диэтилнатриймалоната, однако метод не совсем удачен, так как часто приводит к побочным продуктам, получающимся за счет дегидрогалогенирования алкилгалогенидов. Реакции элиминирования можно до некоторой степени избежать при использовании сопряженного присоединения реактива Гриньяра к алкилиденмалонату, как, например, в синтезе трет-бутилмалоната присоединением метилмагнийиодида к изопропилиденмалонату {схема (23)}. Сопряженное присоединение реактивов Гриньяра к α,β-ненасыщенным сложным эфирам служит основной реакцией; ее можно значительно ускорить в присутствии 1% (мол.) хлорида меди (1). В частности, такие медьорганические реагенты, как LiMeCu и МеСuР(С4Н9-н), селективно присоединяются к β-углеродному атому α,β -ненасыщенных кетонов, обеспечивая потенциальное расширение метода по реакциям, аналогичным приведенным на схеме (23).
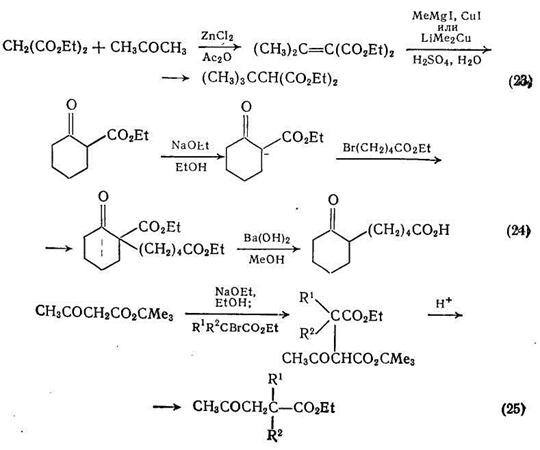
Для получения производных дикарбоновых кислот можно также использовать алкилирование сложных эфиров β-кетокислот {схемы (24) и (25)}. В общем случае продукты этих реакций подвергаются дальнейшим превращениям или, как это показано на схеме (24), используются для получения кетокислот.
Для получения производных сложных эфиров малоновой кислоты можно использовать диэтилоксала, проводя сложноэфирную конденсацию Кляйзена и последующее термическое декарбонилирование {схема (26)}. Это достаточно общий метод введения этоксикарбонильной группы. Применение сложных эфиров, таких, как диэтилсукцинат {схема (27)}, может приводить к получению α-оксопронзводных дикарбоновых кислот путем гидролиза промежуточного сложного эфира β-оксополикарбоновой кислоты.
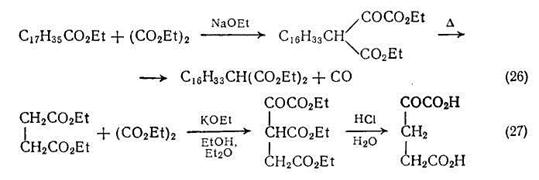
Алкильные производные янтарной кислоты можно получать алкилированием дианиона, в свою очередь полученного из моноэтилсукцината; алкилирование протекает региоспецифично {схема (28)} по соседнему со сложноэфирной группой углеродному атому. Другие а-алкильные производные адипиновой и пимелиновой кислот можно получать более сложной последовательностью реакций {схема (29)}, так как в этом случае анионы легко вступают в циклизацию по Дикману.
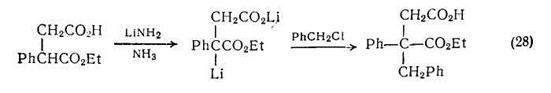
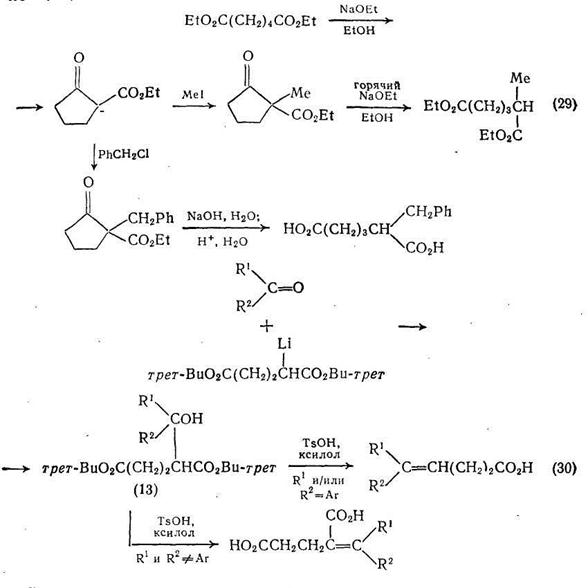
Реакции, аналогичные схеме (28), могут использоваться для синтеза сложных эфиров ненасыщенных дикарбоновых кислот. Например, в результате реакции монолитиевого производного ди-трет-бутилглутарата с различными кетонами с прекрасными выходами получаются сложные эфиры гидроксидикарбоновых кислот (13).
Гидролиз сложных эфиров (13) с одновременной дегидратацией приводит к ненасыщенным производным глутаровой кислоты, если заместители R1 или R2 не ароматической природы {схема (30)}. Однако если один из этих заместителей ароматический, то гидролиз сопровождается не только дегидратацией, но и декарбоксилированием и приводит к ненасыщенным монокарбоновым кислотам.
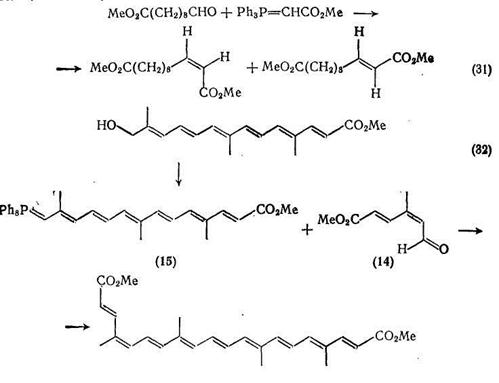
Реакция Виттига — важнейший общий метод региоспецифичного синтеза сложных эфиров α,β -ненасыщенных и полиеновых дикарбоновых кислот. В типичном синтезе {схема (31)} [36], как и во многих подобных случаях, продукт реакции является смесью цис- и транс-изомеров, которые в данном конкретном случае можно разделить дробной кристаллизацией. Особенно широко реакция Виттига применяется в синтезе каротиноидов; в некоторых случаях в этих синтезах используются производные ненасыщенных дикарбоновых кислот. В качестве типичного примера приведем синтез природного биксина {схема (32)}: ключевой интермедиат 5-метоксикарбонил-3-метилпента-цис-2-гранс-4-диеналь (14), как показано на схеме, конденсируется с илидом (15) в стандартных условиях реакции Виттига.
2.3. Реакции Михаэля
Реакция Михаэля используется для получения различных ди- и поликарбоновых кислот. В этом разделе мы рассмотрим несколько типичных примеров этой реакции. Малонат-анион присоединяется к сложным эфирам и нитрилам α,β-ненасыщенных кислот с образованием продуктов, дающих при гидролизе производные глутаровой кислоты {схемы (33)—(36)}.
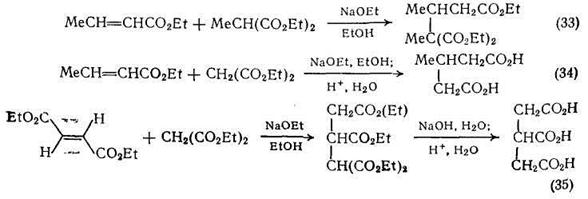
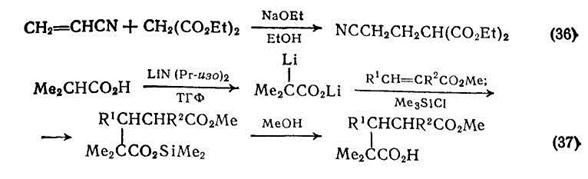
Глутаровые кислоты можно также получить присоединением дианионов карбоновых кислот к α,β-ненасыщенным сложным эфирам {схема (37)}. Дианион изомасляной кислоты получают в тетрагидрофуране при 0°С с использованием двух эквивалентов основания; вслед за присоединением по Михаэлю следует триметилсилилирование продукта.
Полный синтез фунгицида (±)-авенациолида включал в качестве ключевой стадии получение замещенного бислактона (16) в результате сходного с реакцией Михаэля процесса {схема (38)}. На последних стадиях этого синтеза нужная двойная связь вводилась пиролизом сульфоксида в присутствии янтарного ангидрида.
2.4. Окислительные методы
Многие важные пути, ведущие к ди- и поликарбоновым кислотам, включают окисление; некоторые методы нашли практическое применение. Для удобства мы рассмотрим отдельно окисление ароматических и алифатических субстратов.
(1) Получение ароматических кислот
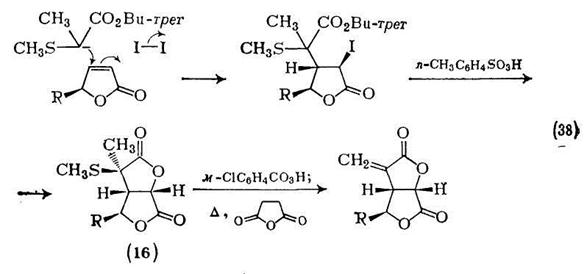
Для получения ароматических ди- и поликарбоновых кислот широко используют окисление боковых цепей различных ароматических соединений. Алкилбензолы, такие как изомерные ксилолы, легко окисляются в соответствующие карбоновые кислоты в жестких условиях. Примеры на схемах (39) —(45) иллюстрируют набор окислительных агентов, которые можно использовать для этой цели.
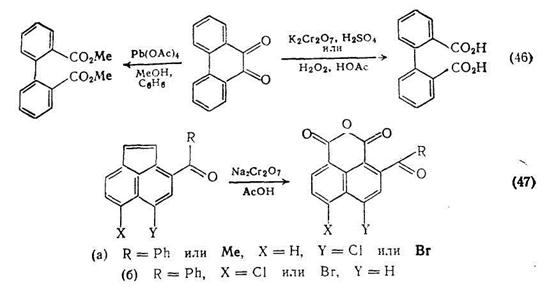
Окисление фенантрахшюна {схема (46)} служит удобным методом синтеза как бифенил-2,2'-дикарбоновой кислоты так и ее диметилового эфира. Окисление различных ацилгалогенаценафтенов приводит к соответствующим нафталиновым аи гидридам, хотя существуют заметные различия в легкости образования ангидридов {схема (47)}.
(2) Получение алифатических кислот
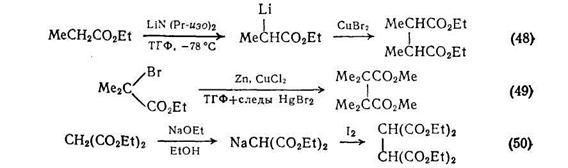
В синтезе дикарбоновых кислот этим путем можно выделить два окислительных процесса: первый включает окислительную димеризацию, второй расщепление углерод-углеродной связи, часто в циклических соединениях {схема (47)}. Сложные эфиры янтарной кислоты можно получать окислительной димеризацией енолят-анионов в присутствии солей меди (II). Метод, использующий литиевые еноляты, {схема (48)} проще и, по-видимому, носит более общий характер, чем альтернативная методика с применением цинкорганических соединений {схема (49)}. Обе реакции напоминают давно известные методы димеризации стабильных анионов, например анионов диэтилмалоната с использованием йода в качестве окислителя {схема (50)}.
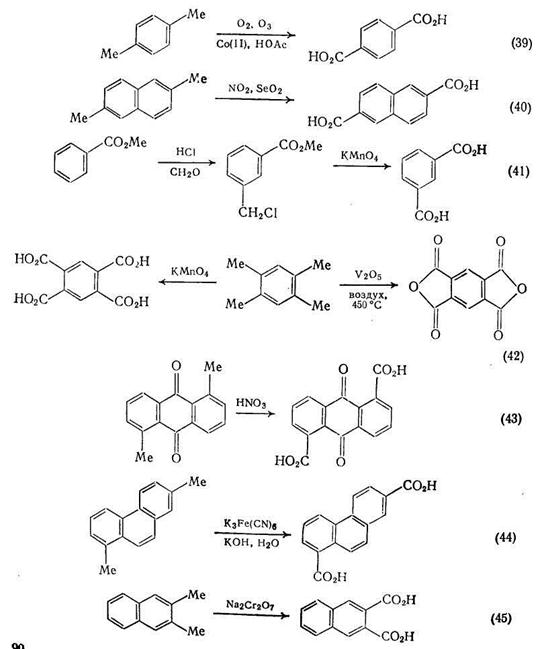
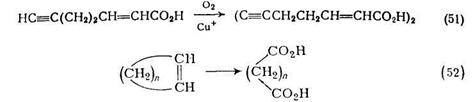
Ацетиленовые кислоты и их эфиры с высоким выходом подвергаются окислительной димеризации в водном этаноле под действием кислорода или воздуха в присутствии хлорида аммония или меди. Эта реакция использована в синтезе кортикроцина контроль за реакцией, которая шла в этом случае с почти количественным выходом при комнатной температуре, осуществлялся по поглощению кислорода {схема (51)}.
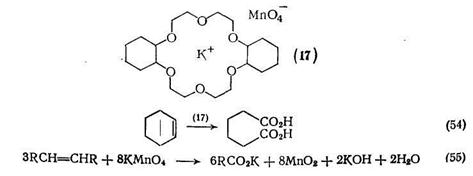
Олефины можно окислять до дикарбоновых кислот {схема (52)} различными способами, и если бы не возникали проблемы, связанные с растворимостью в органических растворителях, наиболее удобным для этой цели был бы перманганат калия. Эти затруднения до некоторой степени преодолимы [49], если использовать в качестве растворителя уксусный ангидрид. Однако в этом случае выходы снижаются, и как показано на примере окисления по схеме (53), могут протекать побочные реакции.

Использование краун-эфиров позволяет снять большинство проблем [50], ибо эти соединения способны образовывать комплексы с солями металлов, что приводит к повышению растворимости в органической среде и повышению реакционной способности анионов. Например, дпцн;слогексил-18-краун-6 образует с перманганатом калия растворимый в бензоле комплекс (17), что дает прекрасный окислитель для органических субстратов. В частности, циклогексен окисляется им с количественным выходом до адипиновой кислоты {схема (54)}. По-видимому, нет оснований предполагать, что механизм этого окисления отличается до такового, действующего в водных средах {схемы (55), (56)}.
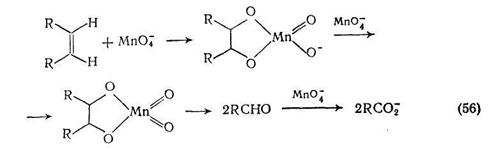
Пля окисления алкенов водным перманганатом калия можно v^nexOM спользовать катализ фазового переноса [51]. Реакции ^Готпатов растворенных в органической фазе, с неорганическими прягентами в водной фазе, которые ингибируются в силу раздела ГД часто катализируются добавлением следовых количеств рас-твпоимых в органической фазе тетраалкиламмониевых или тетра-аакиафосфониевых солей. Предполагают, что катализ осуществляется за счет способности катионов, растворимых в органическом растворителе, многократно переносить анионы в органическую фазу в форме, подходящей для реакции. Этот эффект носит название катализа фазового переноса.
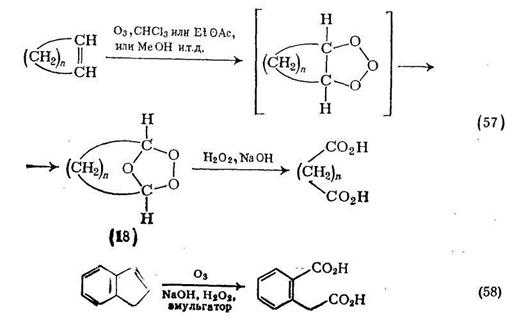
Озонолнз олефинов, как правило, проводят в органических растворителях, часто при низких температурах. Образующийся озонид (18), который обычно слишком нестабилен для безопасного выделения, можно окислять до карбоновых кислот. При окислении циклического олефина продуктом реакции служит дикарбоновая кислота {схема (57)}. Этот двухстадийный процесс можно упростить [52], так как было показано, что в благоприятных случаях эмульсии циклических олефинов и щелочного пероксида водорода мягко реагируют с озоном и с хорошими выходами образуют а,со-дикарбоновые кислоты {схема (58)}.
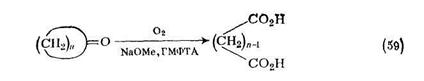
До дикарбоновых кислот можно окислить и другие карбоцикли-ческие соединения. В подходящем растворителе циклические ке-тоны [53] окисляются молекулярным кислородом до дикарбоновых кислот {схема (59)}. Показано, что многие растворители автоокис-ляются в условиях реакции, однако применение гексаметапола (ГМФТА) сводит эти побочные реакции до минимума и позволяет получать удовлетворительные выходы продуктов. Как правило, окисляется наиболее кислая связь С—Н кетона с образованием нестабильного промежуточного перокси-аниона. Полное окисление, аналогичное схеме (59), достигнуто действием азотной кислоты [54J.
Гидролитические методы
Для получения дикарбоновых кислот можно использовать большое число методов, основанных на гидролизе. Часть этих методов мы рассмотрим в данном разделе. Методы, ведущие к сложным эфирам, которые далее можно гидролизовать до ди- или поликар-боновых кислот, рассмотрены в другом месте.
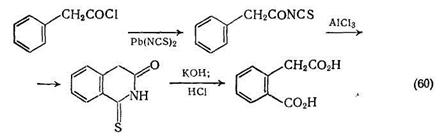
Хотя обычно амиды получают из карбоновых кислот и нитрилов, тиогомофталимиды легко получают из хлорангидридов арил-уксусных кислот [55]. Последующий гидролиз тиогомофталимидов служит удобным методом синтеза гомофталевых кислот {схема (60)}.
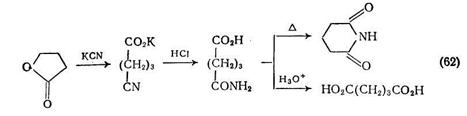
Важным методом синтеза карбоновых кислот является гидролиз нитрилов. В обзоре [56], посвященном малононитрилу, обсуждается его алкоголиз до диэтилмалоната. Синтез янтарной кислоты гидролизом нитрила показан на примере получения [57]; фенилянтарной кислоты {схема (61)}. В этом синтезе использованы конденсация Кневенагеля и последующее присоединение к продукту реакции цианида калия по Михаэлю.
 |
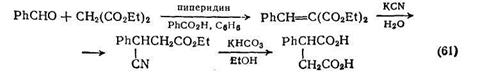
Удобный метод получения глутаровой кислоты [58] основан на гидролизе нитрилов: у-бутиролактон реагирует с цианидом калия при 190 °С, и калиевая соль получающейся цианокислоты гидро-лизуется до глутаровой кислоты. В тщательно контролируемых условиях гидролиза можно выделить моноамид глутаровой кислоты, который затем циклизуется в глутарамид {схема (62)}. Гидролиз динитрилов, получаемых из дигалогенидов, приводит к дикарбоновым кислотам. Дикарбоновая кислота (19), необходимая в качестве интермедиата для синтеза производного пропел-лана [59], была получена именно таким путем {схема (63)}. Подобные реакции замещения цианид-ионом не всегда протекают столь гладко, и как показано на схеме (64), в процессе реакции-замещения промежуточный динитрил (20) циклизуется в енамино-нитрил. Тем не менее гидролиз и размыкание цикла все же приводят к искомой кислоте [59-].
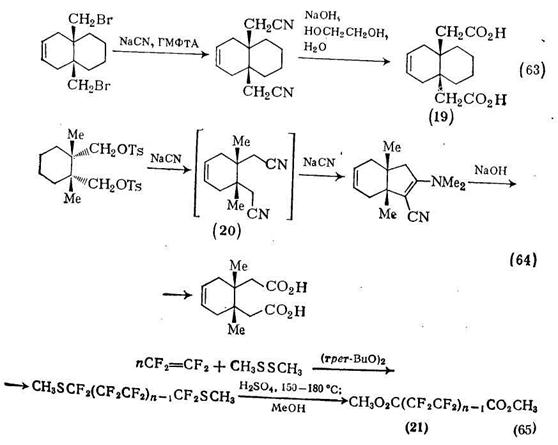
Заслуживает внимания другая методика, включающая гидр0. лиз так как она является общим методом получения перфторал-кандикарбоновых кислот из а,со-бис(метилтио)полифторалканов [60]. Теломеризация тетрафторэтилена в присутствии диметилди-сульфида и грег-бутилпероксида в качестве катализатора приводит к продуктам типа (21) {схема (65)}. Как видно из схемы, эти продукты (п = 2—5) гидролизуются серной кислотой в метаноле до метиловых эфиров фторированных дикарбоновых кислот.
Список литературы
1. Обзор рынка адипиновой кислоты в СНГ. М., ООО «ИНФОМАЙН РЕСЕЧ», 2006, с. 62.
2. Синтезы органических препаратов. Сборник 1. М., ИЛ, 1949.
3. Общая органическая химия. Карбоновые кислоты и их производные. Том 4. М., Химия, 1983, 729с.
4. Богословский Б.Н., Казакова З.С. Скелетные катализаторы, их свойства и применение в органической химии. М., Госхимиздат, 1957.
5. Голодников Г.В. Практические работы по органическому синтезу. Л., Изд-во ЛГУ, 1966, 697с.
6. Губен И., Методы органической химии. Том 2. выпуск 1. М.-Л. Госхимиздат, 1941, 690с
7. Современные методы эксперимента в органической химии. М., Госхимиздат, 1960, 560с.
8. Физер Л., Физер М. Реагенты для органического синтеза. Том 2. М., Мир, 1970, 390.
9. Черонис Н., Микро- и полумикрометоды органической химии. М., ИЛ, 1960, 574.
10. Юрьев Ю.К. Практические работы по органической химии. Выпуск 1 и 2. Изд. 3-е. М., Изд-во МГУ, 1964.
11. Юрьев Ю.К., Левина Р.Я., Шабаров Ю.С., Практические работы по органической химии. Выпуск 4. М. Из-во МГУ, 1969.
12. Шабаров Ю.С. Органическая химия: В 2-х кн. - М.:Химия, 1994.- 848 с.
13. Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. - 623 с.
14. Моррисон Р., Бойд. Органическая химия. - М.: Мир, 1974. - 1132 с.
15. Терней А. Современная органическая химия: В 2 т. - М.: Мир, 1981. - Т.1 - 670 с; Т.2 - 615 с.
16. Робертс Дж., Кассерио М. Основы органической химии: В 2 т. - 2-е изд. -М.: Мир, 1978. - Т.1 - 842 с; Т.2 - 888 с.
17. В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, - 708 с.
18. Фрейдлин Г. Н., Алифатические дикарбоновые кислоты, М., 1978.
© 2009 База Рефератов