
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Курсовая работа: Синтез, свойства и применение дифениламина. Амины и их свойства
Курсовая работа: Синтез, свойства и применение дифениламина. Амины и их свойства
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
СИНТЕЗ ДИФЕНИЛАМИНА
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
Введение
Общие сведения
Применение дифениламина
Обзор литературы. Амины
1. Общие реакции. Образование солей
2. Ацилирование и алкилирование аминов
Ацилирование
Образование производных мочевины
Алкилирование первичных и вторичных аминов
3. Расщепление аминов
4. Окисление аминов
Методы синтеза дифениламина
Синтез на основе анилина и анилиновой соли
Метод взаимодействия нитробензола с бромистым фенилмагнием
Синтез из дихлорбензола
Синтез из хлорбензола и анилина
Список литературы
Общие сведения
Дифениламин (N,N-Дифениламин) – ароматический амин, бесцветное кристаллическое вещество, темнеет на воздухе. Чешуйки или мелкие кристаллы от светло-жёлтого до светло-коричневого или расплав коричневого цвета. Молекулярная масса 169.23; температура кипения 302°C; температура плавления 54-55°C; d425 1.159; d464 1.0513; nd64 1.6189. Хорошо растворяется в диэтиловом эфире, бензоле, ацетоне, CCl4; ограниченно растворяется в этаноле и метаноле; плохо растворим в воде. С минеральными кислотами дифениламин образует соли.
Применение дифениламина
Дифениламин находит широкое применение в различных отраслях; наиболее известные области применения дифениламина:
· полупродукт для синтеза стабилизаторов полимеров, красителей;
· ингибитор окисления и как полупродукт в производстве других ингибиторов окисления для пластичных смазок и при получении лекарственных средств;
· производство тиодифениламина, диафена ФП, N-нитрозодифениламина и для специальных целей.
1. Общие реакции. Образование солей
Амины, являющиеся замещенными производными аммиака, могут быть разделены на три группы: первичные амины общей формулы RNH2, вторичные амины формулы RR'NH и третичные амины, RR'R"N. В этих формулах R соответствует алкильным или арильным радикалам или их замещенным производным, а также ненасыщенным или гетероциклическим радикалам. Кроме того, азот может входить в состав гетероцикла, как, например, и пиридине.
Характер групп, находящихся у атома азота, оказывает большое влияние на основность амина. Обычно алифатические амины, являются сильными основаниями, обладают щелочной реакцией на лакмус и во влажном состоянии поглощают двуокись углерода. Низшие алифатические амины являются, более сильными основаниями, чем аммиак, и титруются кислотами в присутствии метилоранжа или бромфенолблау в качестве индикатора. При наличии ароматического остатка основность аминов выражена значительно слабее; например, анилин и его гомологи, хотя и образуют соли с разбавленными минеральными кислотами, однако не дают щелочной реакции на лакмус и не поглощают двуокись углерода из воздуха. Титрование таких аминов кислотой в присутствии обычных индикаторов не дает удовлетворительных результатов. Напротив, солянокислые соли ароматических аминов легко титруются водным раствором щелочи в присутствии фенолфталеина, т.е. ведут себя в этих условиях, как свободные кислоты. При увеличении числа ароматических радикалов у атома азота наблюдается еще большее уменьшение основности амина. Соли дифениламина гидролизуются в воде в значительной степени с частичным выделением свободного основания. Трифениламин является нейтральным соединением и не образует солей, за исключением комплексного соединения с хлорной кислотой.
Хотя введение нитрогруппы в ядро анилина заметно понижает его основность, нитранилины все же еще обладают основным характером и дают соли с минеральными кислотами, которые, однако, очень легко, гидролизуются при действии воды. Введение более одной нитрогруппы в ядро ведет к дальнейшему понижению основности; действительно, иолинитрамины проявляют лишь незначительную склонность к образованию солей. То же относится и к галоидозамещенным аминам.
Обычно, для получения соли к амину прибавляют небольшой избыток кислоты, например разбавленной серной, концентрированной или разбавленной соляной или бромистоводородной кислоты. Если при этом соль не выпадает, раствор упаривают на водяной бане или в вакуум-эксикаторе до начала кристаллизации. Если галоидоводородные соли плохо кристаллизуются из водного раствора, их получают другим путем, а именно пропусканием сухого галоидоводорода в раствор амина в бензоле, хлороформе или эфире. Этот метод особенно удобен для получения галоидоводородных солей алкиланилинов и диалкиланилинов, а также аминов, соли которых легко гидролизуются водой. При пропускании сухого хлористого водорода в раствор диалкиланилина в сухом эфире до насыщения соответствующие солянокислые соли легко выделяются в кристаллическом состоянии. При этом следует тщательно предохранять реакционную смесь от доступа влаги. Солянокислые соли низших алкиланилинов лучше всего получать в бензольном растворе. Впрочем, в случае алкиланилинов, содержащих сравнительно большие алкильные радикалы, этот способ не дает таких удовлетворительных результатов.
Большинство аминов образует хорошо кристаллизующиеся пикраты, которые могут служить для идентификации аминов или для выделения их из смесей. Обычно, пикраты получаются смешением обоих компонентов в подходящем растворителе, выбор которого определяется сравнительной растворимостью в нем пикриновой кислоты, пикрата и амина. Менее удобно пользоваться для этой цели реакцией обмена. Пикролоновая кислота (I) также применяется для идентификации аминов, особенно в тех случаях, когда пикриновая кислота не дает удовлетворительных результатов. Соли пикролоновой кислоты обычно труднее растворимы, чем пикраты, и обладают более высокой температурой плавления. Этот способ применяется главньм образом для идентификации простейших алифатических производных гидроксиламина, производных морфолина и некоторых алкалоидов 5. Кроме того, для идентификации аминов также применяется имидазолдикарбоновая кислота (II).
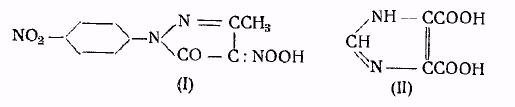
Ароматические амины образуют продукты присоединения с ди- и тринитросоединениями, например с 1, 3, 5-тринитробензолом и 2, 4, 6-тринитротолуолом. Эти продукты также иногда служат для идентификации аминов. Некоторые амины при действии 70%-ной водной хлорной кислоты дают хорошо кристаллизующиеся соли, которые могут служить для их выделения и идентификации.
2. Ацилирование и алкилирование аминов
Третичные амины отличаются от первичных и вторичных аминов отсутствием способных к замещению атомов водорода, связанных с азотом. Это различие ясно проявляется при действии ацилирующих и алкилирующих средств; из первичных и вторичных аминов при ацилировании обычно получаются замещенные амиды, тогда как третичные амины выделяются в неизмененном состоянии после прибавления воды или водной щелочи. Атомы водорода в аминогруппе первичных и вторичных аминов могут быть замещены в определенных условиях алифатическим или ароматическим радикалом, или же остатками –СONH2, —С1, —Вг и —NO2. Эти реакции вкратце рассматриваются ниже.
Ацилирование
Способы, применяемые для ацилирования, могут быть в основном разделены на следующие группы: нагревание аминов с кислотами, взаимодействие аминов с хлорангидридами, бромангидридами или ангидридами кислот и реакция аминов со сложными эфирами, или даже с амидами кислот, дающая обычно худшие результаты.
Первый из этих способов состоит в нагревании амина с избытком соответствующей карбоновой кислоты.
![]()
Аналогичным путем получаются высшие гомологи ацетанилида. Этот способ часто применяется для идентификации одноосновных кислот. Интересно отметить, что муравьиная кислота значительно легче, чем ее гомологи, превращается в замещенные формамиды по этому способу. Форманилид легко образуется при нагревании 50%-ной водной муравьиной кислоты с анилином.
Для ацетилирования аминов рекомендуется также пользоваться тиоуксусной кислотой. Преимущество этого способа состоит в том, что ацетилирование анилина и его гомологов протекает в этом случае на холоду. Реакция протекает с выделением сероводорода.
![]()
Более удобный и распространенный способ получения ацилированных аминов заключается в применении хлорангидридов или ангидридов кислот. Хлорангидрид кислоты реагирует с избытком амина с образованием ацилированного производного и солянокислой соли амина.
![]()
Отделение солянокислой соли от ацилированного производного амина основано на их различной растворимости. Обычно реакцию ведут в таком растворителе, в котором соль амина нерастворима. Кроме того, если ацилированный амин нерастворим в воде, солянокислую соль легко удалить промыванием реакционной смеси водой
Если хлорангидрид кислоты сравнительно устойчив к действию воды и холодного водного раствора щелочи, введение ацильной группы может быть осуществлено по способу Шоттена и Баумана. Амин суспендируют в приблизительно 10%-ном водном растворе щелочи и обрабатывают хлорангидридом кислоты, взятым в 1,25—1,5-кратном против теории количестве. При этом реакционную смесь перемешивают или взбалтывают, пока большая часть хлорангидрида не прореагирует. Избыток хлорангидрида разлагают слабым нагреванием реакционной смеси. Образовавшееся трудно растворимое ацильное производное отфильтровывают, промывают водой до полного удаления щелочи и перекристаллизовывают из подходящего растворителя. Важно, чтобы в процессе реакции водный раствор-все время обладал щелочной реакцией. Этот метод с успехом применяется для хлорангидридов ароматических кислот, арилсульфоновых кислот и пирослизевой кислоты. Следует отметить, что сульфонильные производные первичных аминов растворимы в щелочах, а сульфонильные производные вторичных аминов нерастворимы. На этом основан способ распознавания и разделения первичных и вторичных аминов.
К другим способам ацилирования относятся действие хлорангидрида кислоты на эфирный раствор амина с суспендированным в нем углекислым калием, а также ацилирование в пиридине.
Для ацетилирования первичных ароматических аминов в лабораторных условиях целесообразно пользоваться уксусным ангидридом. Реакция между уксусным ангидридом и анилином или его гомологами протекает очень легко и обычно осуществляется прибавлением уксусного ангидрида к смеси амина примерно с 5-кратным по объему количеством воды. При реакции происходит выделение тепла и смесь быстро густеет, вследствие выделения ацетильного производного. Если амин обладает сравнительно высоким молекулярным весом, обычно удобнее перед прибавлением уксусного ангидрида смешивать основание с разбавленной уксусной кислотой. Применение спирта в качестве растворителя при ацетилировании аминов уксусным ангидридом на холоду имеет то преимущество, что избыток ангидрида легко можно удалить 1—2-кратным выпариванием со спиртом. Ацетилирование уксусным ангидридом в водной или спиртовой среде не дает удовлетворительных результатов при первичных ароматических аминах, содержащих в ядре отрицательные заместители.
Вышеупомянутый способ ацетилирования обладает тем преимуществом, что при этом не наблюдается образования диацетильных производных, что имеет место при применении неразбавленного уксусного ангидрида, например, в результате нагревания 10 г анилина с 40 г уксусного ангидрида в течение 1 часа продукт реакции представляет собой смесь, содержащую 10 г диацетиланилина и 5,6 г ацетанилида18. Наличие заместителей, например СНз, —NO2, —С1, —Вг, в о-положении к аминогруппе благоприятствует образованию диацетильных производных. При нагревании о-толуидина с 4-кратным по весу количеством уксусного ангидрида с обратным холодильником получается диацетил-о-толуидин с прекрасным выходом.
Наличие в ядре ароматического амина нитрогруппы и, в меньшей степени, хлора или брома замедляет реакцию ацетилирования при комнатной температуре. Это явление становится особенно заметным при накоплении отрицательных групп в молекуле. При стоянии раствора 2, 4, б-триброманилина в избытке уксусного ангидрида при комнатной температуре в течение 2 недель ацетильное производное не образуется. Присутствие небольшого количества концентрированной серной кислоты очень сильно катализирует процесс ацетилирования, из 1 в 2, 4, 6-триброманилина в 20 г уксусного ангидрида в присутствии двух капель концентрированной серной кислоты при стоянии в течение 10 мин. при комнатной температуре получается чистый 2, 4, 6-трибромацетанилид. Для выделения продукта реакционную смесь выливают в воду.
Наилучший способ получения ацетильных производных низших алкнланилинов состоит в перегонке смеси равных объемов амина и уксусного ангидрида; выше 200° перегоняется ацетильное производное в довольно чистом состоянии. Многие из этих соединений кристаллизуются при охлаждении.
Третичные амины, благодаря своему строению, не способны к образованию амидов при взаимодействии с хлорангидридами или ангидридами кислот. Однако они могут давать с хлорангидридами продукты присоединения, которые обычно разлагаются при действии воды с образованием исходного амина. Например, продукт присоединения 1 моля пиридина к 1 молю хлористого ацетила при действии спирта превращается в солянокислый пиридин и уксусноэтиловый эфир. Хлористый диаллил также образует продукт присоединения к пиридину. Кроме того, описаны соединения, образующиеся при взаимодействии хлористого бензоила и хлористого ацетила с триэтиламином, пиридином, диметиланилином и некоторыми другими третичными аминами. Продукты присоединения триметиламина к арилсульфохлоридам сравнительно стойки к действию воды и дают хлороплатинаты и хлораураты.
Образование производных мочевины
Соли первичных и вторичных аминов с циановой кислотой более или менее легко изомеризуются с образованием замещенных производных мочевины
RNH2 HCNO -—> RNHCONH2
Эта реакция аналогична превращению циановокислого аммония в мочевину. Следующие примеры иллюстрируют применение этого способа. Первичные и вторичные амины легко реагируют с эфирами изоциановой кислоты с образованием производных мочевины. Обычно для этой цели применяют фенилизоцианат, который нагревают с эквимолекулярным количеством амина в каком-либо не содержащем гидроксила растворителе, например в петролейном эфире.
![]()
Совершенно необходимо предохранять реакционную смесь от доступа влаги и применять лишь тщательно обезвоженные растворитель и амин, так как фенилизоцианат реагирует с водой с образованием дифенилмочевины. α-Нафтилизоцианат более удобен в этом отношении, чем фенилизоцианат, так как он менее чувствителен к действию воды и поэтому при реакции образуется меньшее количество нежелательных побочных продуктов.
Фенилизотиоцианат (фенилгорчичное масло) реагирует с аминами аналогичным образом с образованием соответственных производных тиомочевины. Реакция эта осуществляется в тех же условиях, как и с фенилизоцианатом.
Алкилирование первичных и вторичных аминов
Последовательное замещение алкильными группами атомов водорода, находящихся у азота в первичных аминах, ведет к образованию вторичных и третичных аминов. Введение алкильных групп легко достигается действием на амин соответственного галоидного алкила или алкилсульфата. Состав конечного продукта реакции зависит в значительной степени от относительных количеств взятых в реакцию компонентов, а также и от условий опыта, и обычно очень трудно получить при алкилировании только одно из возможных производных амина и поэтому продукт реакции, как правило, представляет собой смесь вторичного и третичного аминов наряду с значительным количеством непрореагировавшего первичного амина, а часто с примесью некоторого количества соли четвертичного аммониевого основания. Получение сложной смеси при применении галоидного алкила является результатом образования при реакции галоидоводорода, который дает соли с находящимися в реакционной смеси аминами. Распределение галоидоводорода между аминами зависит от их относительной основности, их сравнительного количества, а также от растворимости солей аминов в реакционной смеси. При алкилировании ароматических аминов выделяющийся осадок обычно содержит значительное количество соли исходного амина, а в растворе остается алкилированный амин, который вступает в дальнейшую реакцию с галоидным алкилом. Такие затруднения могут быть преодолены, по крайней мере, до известной степени проведением алкилирования в присутствии веществ, способных связывать образующийся галоидозодород, например, углекислой или двууглекислой соли щелочного металла. Для выделения вторичных аминов, наряду с вышеупомянутыми способами, обычно, за исключением некоторых особых случаев, пользуются способностью вторичных аминов образовывать нитрозамины. При восстановлении нитрозаминов оловом с соляной кислотой или при нагревании их с минеральными кислотами получаются чистые вторичные амины. Другой способ, по которому удается получать вторичные амины с значительно лучшими выходами, основан на способности металлических производных многих замещенных амидов типа RCONHR реагировать с галоидными алкилами. Из продукта алкилирования при гидролизе получается вторичный амин
![]()
Для этой цели удобно пользоваться ацетанилидом и его гомологами. Кроме того, применялись и формильные производные первичных ароматических аминов, а также арилсульфонильные производные первичных аминов.
Другой способ получения гомологов метиланилина заключается в нагревании галоидного алкила с большим избытком ароматического амина. По окончании реакции избыток ароматического амина осаждают прибавлением водного раствора хлористого цинка. Этот способ применялся для получения многих алкиланилинов с вполне удовлетворительными результатами. Таким же путем могут быть получены алкиланилины, содержащие третичную алкильную группу. Для алкилирования аминов также можно пользоваться ди-алкилсульфатами. Впрочем, обычно, этот способ ограничивается применением имеющегося в продаже диметилсульфата. Алкилирование по этому способу проводится в индиферентном растворителе или в присутствии водной щелочи, причем последняя модификация имеет более широкое применение. Вместо диалкилсульфатов можно1 применять эфиры арилсульфоновых кислот. Спирты вступают в реакцию с солями первичных ароматических аминов, примерно, при 200° с образованием моно- и диалкилариламинов. Эта реакция имеет применение в промышленности; для получения метиланилина нагревают при 180° смесь 55 частей солянокислого анилина и 16 частей метилового спирта. Для получения диметиланилина смесь 80 частей анилина, 78 частей метилового спирта и 8 частей серной кислоты нагревают в автоклаве до 235°. В лабораторных условиях можно вместо серной кислоты пользоваться другим катализатором, например йодом. Еще более активным катализатором в этой реакции является смесь порошкообразной меди с бромистым натрием или смесь галоидных солей меди и натрия Вторичные амины могут быть также получены восстановлением. Эта реакция может быть осуществлена электролитическим путем, действием цинковой пыли и водной щелочи натрия в спиртовой среде или муравьиной кислоте.
Новый способ получения метальных производных α- и β-нафтиламинов предложен Родионовым и Введенским. Для получения моно- и диметильных производных применяется действие метилового эфира р-толуолсульфоновой кислоты на соответствующий амин.
Другой интересный способ получения вторичных аминов основан на взаимодействии азометинов с йодистыми алкилами, причем образуются соединения, которые по прибавлении воды или спирта расщепляются на вторичный амин и альдегид.
![]()
Арилирование первичных и вторичных аминов
Введение в аминогруппу ароматического остатка обычно сопряжено с некоторыми затруднениями, вследствие малой реакционноспособности галоида в ароматических соединениях. Например, хлорбензол и бромбензол не вступают в реакцию с анилином в условиях, аналогичных применяемым для получения этиланилина. Впрочем, в присутствии медной бронзы или йодистой меди эта реакция протекает более гладко.
При взаимодействии третичных аминов с йодистыми алкилами образуются соли четвертичных аммониевых оснований. Общий способ получения таких соединений состоит в смешении обоих компонентов, иногда в каком-либо подходящем растворителе. Реакция протекает при комнатной температуре или при нагревании по схеме.
R'"R"R'N + RHal —> R'"R"R'RNHal
Вместо галоидных алкилов можно пользоваться диалкилсульфатами или алкиловыми эфирами ароматических сульфоновых кислот, причем получаются сернокислые или арилсульфоновокислые соли соответствующих четвертичных аммониевых оснований.
Реакция образования солей четвертичных аммониевых оснований часто применяется для идентификации третичных аминов, причем в качестве реактива наибольшее применение имеет йодистый метил. Рекомендуется также пользоваться для этой цели метиловым эфиром р-толуолсульфоновой кислоты. Ниже приводятся общие условия реакции для получения р-толуол-сульфоновокислых солей четвертичных аммониевых оснований.
При взаимодействии третичных аминов с йодистыми алкилами образуются соли четвертичных аммониевых оснований. Общий способ получения таких соединений состоит в смешении обоих компонентов, иногда в каком-либо подходящем растворителе. Реакция протекает при комнатной температуре или при нагревании по схеме
R'"R"R'N + RHal —> R'"R"R'RNHal
Вместо галоидных алкилов можно пользоваться диалкилсульфатами или алкиловыми эфирами ароматических сульфоновых кислот, причем получаются сернокислые или арилсульфоновокислые соли соответствующих четвертичных аммониевых оснований.
Соли четвертичных аммониевых оснований образуются не только в результате взаимодействия галоидных алкилов или эфиров ароматических сульфоновых кислот с третичными амидами, но и при действии эфиров йодуксусной кислоты на некоторые амины. Легче других вступают в эту реакцию бензилпиперидин, алифатические третичные амины и хинолин. В некоторых случаях для получения четвертичных аммониевых солей.
Легкость образования четвертичных аммониевых солеи сильно зависит от характера исходных соединений.
Наличие заместителей в о-положении к аминогруппе оказывает замедляющее действие на скорость реакции, что ясно видно из сравнения констант, для диметил-о-, -т- и р-толуидина, а также для хинолина и изохинолина. Это явление еще более резко выражено при наличии двух заместителей в о-положении к аминогруппе.
Например, третичные амины (III) и (IV) не реагируют с йодистым метилом при 100°, тогда как изомерные им амины, обладающие другим строением, сравнительно легко образуют четвертичные аммониевые соли
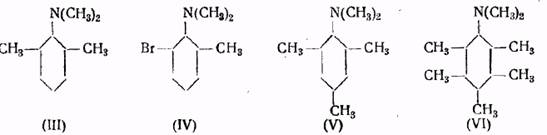
Диметилмезидин (V) и диметиламинопентаметилбензол (VI) также не способны к образованию четвертичных аммониевых соединений
Один из наиболее старых методов расщепления аминов заключается в нагревании сухой галоидоводородной соли алкиламина. В строго определенных условиях, зависящих главным образом от характера исследуемого вещества, удается отщепить одну или несколько алкильных групп и тем самым превратить третичный амин последовательно во вторичный, затем в первичный амин и, наконец, в аммиак. Например, при нагревании солянокислого триметиламина при 285° образуется смесь солянокислого диметиламина и свободного триметиламина с выделением хлористого метила. При более высокой температуре продукт реакции может содержать метиламин или хлористый аммоний. В случае высших алифатических аминов этот способ не получил применения. Более широкое применение он имеет для расщепления алкилариламинов; при нагревании метиланилина в слабом токе хлористого водорода образуются солянокислый анилин и хлористый метил. Аналогично ведет себя трибензиламин. При сухой перегонке солянокислого диметиланилина получаются хлористый метил, солянокислый анилин и метиланилин. Бромистоводородные соли моноалкилпроизводных анилина, от этиланилина до изоамиланилина, при нагревании в пределах от 250 до 300° разлагаются с отщеплением алкильной группы в виде смеси олефина и бромистого алкила. В этих условиях образование р-замещенных аминоалкилбензолов, вследствие перемещения алкильной группы в ядро, происходит лишь в незначительной степени. Если реакцию вести в закрытом сосуде, чтобы воспрепятствовать улетучиванию продуктов реакции, выход р-аминоалкилбензола увеличивается за счет уменьшения количества получающихся летучих продуктов. Эта перегруппировка впервые наблюдалась Гофманом.
Алкиланилины, содержащие у азота третичные алкильные группы, сравнительно легко их отщепляют; уже при нагревании таких алкиланилинов с водными минеральными кислотами до 110—140° в течение нескольких часов третичные алкильные группы отщепляются в очень значительной степени.
Интересно, что разложение солей аминов, в частности фосфорнокислых солей, может применяться для получения олефинов и, в особенности, диенов. По этому способу из у-амино-а-фенилбутана получается смесь фенилбутенов, из диаминометилпентана образуется метилпентадиен, а 1,3-диаминоцикло-гексан превращается при этом в 1,3-циклогексадиен. Этот способ может также быть использован для получения пентадиенов.
Другой тип расщепления наблюдается при действии тетранитрометана на третичные амины, причем образуются соответственные нитрозамины.
![]()
Диалкилариламины лучше всего реагируют с тетранитрометаном в присутствии пиридина, а алифатические амины — в уксусной кислоте. Общие условия расщепления диалкилариламинов по этому способу заключаются. Другой, менее общий способ расщепления аминов, основан на действии хлорноватистой кислоты или хлора на водный раствор алифатического третичного амина или циклического амина, например тропидина.
По мнению Мейзенхеймера реакция протекает следующим образом:
![]()
Для отщепления одной алкильной группы от третичных аминов обычно пользуются реакцией с бромистым цианом. Реакция эта протекает по уравнению
![]()
У третичных аминов, являющихся производными пиперидина, тетрагидрохинолина или дигидроизоиндола, может происходить либо нормальное образование цианамидов, либо размыкание цикла.
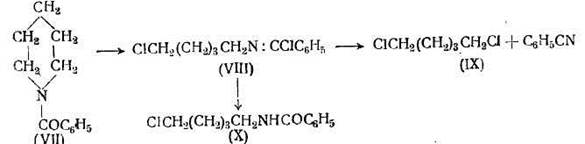
Более удовлетворительный способ размыкания цикла некоторых циклических аминов заключается в нагревании соответственных бензоильных производных с пятихлористым фосфором. Например, бензоилпиперидин (VII) гладко превращается в этих условиях в соединение (VIII), которое при гидролизе дает бензоиламино-ξ-хлорпентан (X) и дихлорпентан (IX)
При окислении первичных ароматических аминов образуются в зависимости от условий различные продукты.
При окислении первичных ароматических аминов кислотой Каро аминогруппа превращается в нитрозогруппу. У аминов жирного ряда процесс протекает более сложно, так как первоначально образующиеся продукты окисляются еще легче, чем исходные амины. При действии кислоты Каро на этиламин образуются наряду с основным продуктом реакции — уксусной кислотой — небольшие количества нитроэтана, ацетгидроксамовой кислоты, ацетоксима и ацетонитрила. Более удовлетворительные результаты получаются, если аминогруппа присоединена к третичному атому углерода. Например, из третичного бутиламина и третичного амиламина последовательно образуются соответственные алкилгидроксиламины и алкилнитрозосоединения. Ароматические амины гладко окисляются в соответственные нитрозосоединения
![]()
Эта реакция является довольно важной, так как представляет удобный способ получения нитрозопроизводных ароматических соединений. При дальнейшем окислении полученного таким образом нитрозосоединения образуется нитросоединение, причем этот способ введения нитрогруппы в ароматическое ядро применяется на практике в тех случаях, когда обычный метод нитрования оказывается непригодным.
При применении других окислителей реакция может быть направлена в сторону образования иных продуктов. Так, из анилина можно при определенных условиях получить азобензол и азоксибензол, фенилхинондиимин C6H5N:С6H4:NH, эмеральдин, черный анилин и р-бензохинон. При окислении в мягких условиях, а именно при действии перекиси свинца в уксуснокислой среде на эфирный раствор анилина образуются азобензол и фенилхинондиимин. Имеющиеся к настоящему времени опытные данные приводят к выводу, что образование сложных продуктов окисления анилина — эмеральдина и черного анилина — связано с полимеризацией фенилхинондиимина или его взаимодействием с непрореагировавшим анилином. По-видимому, получению р-бензохинона при окислении анилина хромовой кислотой в водном растворе предшествует образование черного анилина. Действительно, при окислении черного анилина двухромовокислой солью в разбавленной серной кислоте образуется р-бензохинон с выходом около 85% от теоретического; при применении в качестве окислителя перекиси свинца и серной кислоты выход р-бензохинона достигает 95%.
Этот способ пригоден также для получения гомологов р-бензохинона. Например, из о- или т-толуидина образуется р-толухинон, из р-ксилидина — р-ксилохинон. При окислении некоторых аминов, содержащих метильную группу в р-положении к аминогруппе, эта метальная группа отщепляется и образуются р-хиноны; из мезидина образуется 2,6-диметил-р-бензохинон, из псевдокумидина получается р-ксилохинон.
Методы синтеза дифениламина
Синтез на основе анилина и анилиновой соли
93 г анилина и 93г солянокислого анилина (анилиновой соли) нагревают в течение 20 час. при 230° в эмалированном автоклаве с эмалированной гильзой для термометра. Давление поднимается до 6 атм. Если не имеется эмалированной гильзы для термометра, то можно просто нагревать при указанном давлении, следя за температурой масляной бани. Она примерно и а 25° выше, чем действительная температура внутри автоклава. .. Через два часа осторожно спускают через вентиль воду, так как даже следы ее вредно сказываются на течении реакции. Продувание повторяют три раза в течение часа, при этом выделяются также немного анилина и аммиак. Нагревать более 20 час не имеет смысла, так как при этом выход может только уменьшиться. После охлаждения содержимое автоклава переносят в фарфоровую чашку и прибавляют 1 л воды. Затем нагревают до 800С, подкисляют 30%-иой соляной кислотой до кислой реакции на конго и оставляют опять охлаждаться на ночь. Неочищенный дифениламин выделяется в виде твердой лепешки, которую можно легко отделить от маточного раствора, так как дифениламин не образует соли с разбавленной соляной кислотой. Отфильтрованный дифениламин расплавляют еще раз с некоторым количеством воды, экстрагируют небольшим количеством соляной кислоты и промывают разбавленным раствором соды.
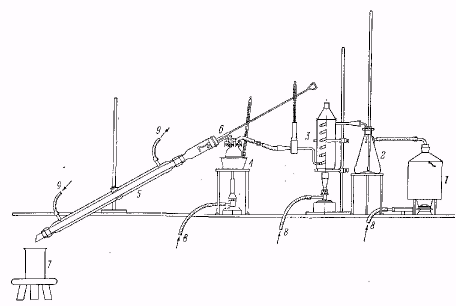
Лабораторная установка для перегонки с перегретым водяным паром.
1 – парообразователь, 2 – водоотделитель, 3 – пароперегреватель, 4 – перегонный сосуд в масляной бане, 5 – холодильник с широкой трубкой. 6 – приспособление для проталкивания твердого вещества, застрявшего в холодильнике, 7 – приемник для дистиллята, 8 – газовая горелка, 9 – водяное охлаждение.
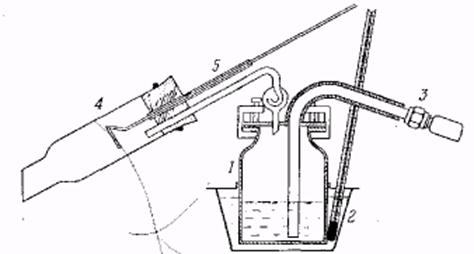
Лабораторный сосуд для перегонки с перегретым водяным паром. 1 – медный куб, 2 – масляная баня с термометром, 3 – соединение спароперегревателем, 4 – холодильник, 5 приспособ для проталкивания.
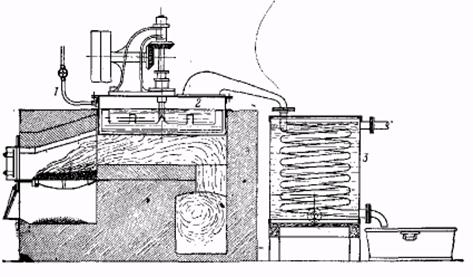
Производственная установка для перегонки с перегретым водяным паром а-нафтиламина, дифениламина и т. д. 1 – ввод перегретого водяного пара, 2 – перегонный сосуд с медленно вращающейся мешалкой, 3 – змеевиковый холодильник.
Полученный таким образом дифениламин очень загрязнен. Поэтому его следует перегнать с перегретым водяным паром. Для этого собирают аппаратуру, как показано на рисунках; перегонный сосуд берут емкостью примерно 500 мл. Масляную баню нагревают до 250° и затем приводят в действие перегреватель на обычной печи Флетчера. Пар должен быть тщательно высушен и перегрет до 300°. При хорошей перегонке легко удается с одной частью поды отогнать 0,5 части основания. Дифениламин получают в виде почти бесцветной жидкости, застывающей в бледно-желтую лепешку. После сливания поды получают около 100 г совершенно чистого дифениламина с т. пл. 51°.
Из кислого маточного раствора можно регенерировать около 35 г анилина.
Технические примечания. Должны быть эмалированы ни только нижняя часть, но и крышка автоклава. Следы железа или меди снижают выход дифениламина на 30—35% и вызывают осмоление. Экстракцию соляной кислотой проводят в деревянных чанах; перегонку с перегретым водяным паром осуществляют в приборе, изображением на рис. Для перегревания водяного пара применяют новые аппараты, например так называемый пароперегреватель Гейцмана и др. С одной частью воды можно перегонять при 2300 до одной части дифениламина.
Метод взаимодействия нитробензола с бромистым фенилмагнием
Действие нитробензола на бромистый фенилмагний. К бромистому фенилмагнию, полученному из 78,5 г бромбензола (0,5 моля) и 12 г магния, по каплям прибавлено 15,4 г нитробензола, разбавленного восьмикратным объемом абсолютного эфира. При этом реакционная смесь часто встряхивалась и охлаждалась. Через 15 мин. после прибавления всего нитробензола продукт реакции осторожно разложен льдом и к смеси прилита соляная кислота до сильно кислой реакции. Фенол из эфирного раствора многократно извлечен 10%-ным КОН; при добавлении к щелочному раствору хлористого натрия и соляной кислоты выделено 7,4 г фенола (выход 63%). Из эфирного раствора после отгонки эфира и перегонки в вакууме выделено 9,6 г дифенила (выход 50%) и 13,3 г дифениламина (выход 63%).
При взаимодействии тринитробензола с RMgBr (где R = алкил) после разложения водной уксусной кислотой выделен 1,3,5-триалкил-2,4,6-тринитроциклогексан.
Синтез из дихлорбензола
Получение р-фенилендиамина из р-дихлорбензола. 1,00 частей дихлорбензола, 750 частей аммиака и 20 частей медного купороса нагревают в автоклаве около 20 час при 170-180° и, под конец, при 200°. Избыточный аммиак отгоняют и р-фенилендиамин изолируют в форме хлоргидрата или сульфата или в виде свободного основания.
При действии водного аммиака на хлорбензол в присутствии медных соединений при высокой температуре и давлении получаются анилин, фенол и дифениламин. При оптимальных условиях (1 моль хлорбензола + 5 молей NH3 в виде 30—34%-ного водного раствора 0,2 моля Сu2О при 70 от и 200—230° и при 80%-ном наполнении автоклава) реакция заканчивается в 2—3 часа. Выход составляет 89—90% анилина. 4,5% фенола и I—2% дифениламина. Выход анилина увеличивается с увеличением концентрации и общего количества аммиака, понижением температуры и увеличением степени наполнения автоклава.
Синтез из хлорбензола и анилина
Карпухин получил дифениламин по уравнению
![]()
Получение дифениламина из анилина и хлорбензола. 9 частей анилина, 18 частей хлорбензола и 14 частей натронной извести нагревают при 280° в течение 5 часов в запаянных стеклянных трубках в автоклаве, наполненном ксилолом (чтобы трубки не испытывали сильного давления, так как упругость паров хлорбензола и ксилола почти одинакова). Выход дифениламина 10,2%.
При увеличении температуры и времени нагревания выход повышается, но одновременно увеличивается потеря анилина от разложения.
При охлаждении массу разбавляют водой и нейтрализуют соляной кислотой до слабокислой реакции (проба на конго), при этом анилин переходит в раствор, а дифениламин выделяется в виде масла. На другой день затвердевший дифениламин отфильтровывают, промывают и сушат. Сырой дифениламин очищают перегонкой е перегретым паром.
Второй способ очистки: дифениламин перегоняют с двукратным количеством нефтяного погопа, кипящего при 250—315°, причем он увлекается парами нефтяных погопов и частично в них растворяется. Затем пропускают хлористый водород и получают хлористоводородную соль дифениламина в виде белых кристаллов, которые отфильтровывают и промывают легким бензином. Анилин, не вошедший в реакцию, может быть регенерирован из раствора его солянокислой соли.
Синтез из хлорбензола и анилина
Гораздо большее практическое значение имеет замена гидроксила аминогруппой в ароматическом ряду, чем в алифатическом. Для фенолов с этой целью также прибегают к двойному соединению хлористого цинка и аммиака. Тем не менее опыты превращения фенола в анилин со спиртовым или водным аммиаком приводят к чрезвычайно незначительным выходам. С двойным соединением хлористого цинка и аммиака получают уже при 300° значительное количество анилина и дифениламина, а при 350° около 70% (веса фенола) обоих оснований. Особенно подходящей является температура 3300. Главная реакция полностью протекает за 20 час.
Присутствие хлористого аммония и большого, избытка двойного соединения хлористого цинка и аммиака весьма влияет на выход.
Чем меньше ZnCl2•2NH3 и NH4Cl употребляется, тем больше образуется дифениламина вместо анилина, особенно если нагревание происходит долго и при более высокой температуре. Так как дифениламин при нагревании с двойным соединением хлористого цинка и аммиака в присутствии хлористого аммония образует анилин, то надо считать механизм образования последнего ясным.
Аналогично действию хлористого аммония и двойного соединения хлористого цинка с аммиаком реагируют соответствующие соединения брома.
Получение анилина из фенола при помощи хлорциикаммиака и хлористого аммония. 10 г фенола, 40г ZnCl2•2NH3 и 4 г NH4C1 нагревают под давлением в трубке в течение 20 часов при 330°. Содержимое трубки растворяют при нагреваний в соляной кислоте до образования двух слоев. Фенол и дифениламин экстрагируют эфиром, из солянокислого раствора анилин осаждают аммиаком и извлекают эфиром. Эфирный раствор фенола и дифениламина обрабатывают щелочью для освобождения от фенола. Таким образом, получают, считая на взятый фенол, около 44% анилина, 38% дифениламина и 6% свободного фенола.
Список литературы
1. Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. - 623 с.
2. Терней А. Современная органическая химия: В 2 т. - М.: Мир, 1981. - Т.1 - 670 с; Т.2 - 615 с.
3. В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, - 708 с.
4. Шабаров Ю.С. Органическая химия: В 2-х кн. - М.:Химия, 1994.- 848 с.
5. Губен И. Методы органической химии. Том 4, выпуск 1, книга первая, М.-Л., Госхимиздат, 1949, 894с.
6. Фирц-Давид Г.Э. Производство органических красок. М.-Л., Госхимтехиздат, 1933, 633с.
7. Фирц-Давид Г.Э., Бланже Л. Основные процессы синтеза красителей. М., ИЛ, 1957, 584с.
8. Хиккинботтом В. Реакция органических соединений. М., ГОНТИ, 1939, 556с.
9. Иоффе С.Т., Несмеянов А.Н. Методы элементоорганической химии (магний, бериллий, кальций, стронций, барий). М., Изд-во АН СССР, 1963, 788с.
© 2009 База Рефератов