
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Курсовая работа: Синтез хлороформа. Реакции нуклеофильного замещения и элиминирования галогеналканов
Курсовая работа: Синтез хлороформа. Реакции нуклеофильного замещения и элиминирования галогеналканов
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ ХЛОРОФОРМА”
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
1. Введение
1.1. Свойства хлороформа
1.2. Применение хлороформа
1.3. Меры предосторожности при работе с йодоформом
1.4. Синтезы йодоформа
1.4.1. На основе карбоната натрия, йода и этилового спирта [1]
1.4.2. На основе ацетона, йода и щелочи [2]
1.4.3. Электролитический способ [3]
1.4.4. На основе ацетона, йодистого калия, гипохлорита калия, этилового спирта [4]
1.4.5. Промышленная методика производства йодоформа [5]
2. Литературный обзор
2.1. Реакции нуклеофильного замещения галогеналканов
2.1.1. Бимолекулярное нуклеофильное замещение
2.1.2. Мономолекулярное нуклеофильное замещение
2.1.3. Сравнение реакций SN 1 и SN 2
2.2. Реакции отщепления (элиминирование)
2.2.1. Бимолекулярное отщепление Е2
2.2.2. Мономолекулярное отщепление Е1
2.2.3. Сравнение реакций нуклеофильного замещения и элиминирования
2.3. Методы синтеза галогеналканов
2.3.1. Галогенирование алканов
2.3.2. Присоединение галогенводородов к олефинам
2.3.3. Замещение гидроксильной группы спиртов на галоген
3. Методика эксперимента
4. Выводы
Список литературы
1. Введение
1.1. Свойства хлороформа
Йодоформ (CHI3) – мелкокристаллический порошок лимонного цвета, практически нерастворимый в воде, легко растворимый в эфире, хлороформе, мало растворимый в спирте, бензине, органических маслах. Температура плавления кристаллов йодоформа 1190С.
Химические свойства йодоформа не отличаются от свойств остальных галогеналканов, и подробно рассмотрены ниже; они обусловлены спецификой строения.
Вследствие высокой электроотрицательности галогена связи углерод-галоген являются сильно полярными Сd+®Id-. Естественно, атомы йода, связанные с электронодефицитным углеродом, можно легко заменить на частицу, богатую электронами, реагент – нуклеофил Nu. Кроме того, характерной для галогеналканов, в частности, для йодоформа, является реакция отщепления (элиминирования). Помимо этого, галогеналканы образуют магнийорганические соединения R-MgX, чрезвычайно важные в синтетическом отношении.
В продаже существуют следующие сорта йодоформа:
1. Iodoform crystallisatum. Твердые кристаллы. Получают его перекристаллизацией обычного йодоформа из спирта. Для этого берут отходы производства других сортов, отсев, сметки и т.д. Кристаллический йодоформ применяется редко, преимущественно в Англии и англоговорящих странах.
2. Iodoform pulvis levissium. Представляет собой смесь мелкого порошка и нежных листочкообразных кристаллов. При этом сорте главное значение имеет легкость порошка.
3. Iodoform farinosum. Мелкие очень легкие листочкообразные кристаллы с небольшим количеством пороша.
4. Iodoform pulvis subtilis. Светложелтый порошок почти без кристаллов. Он тяжелее, чем другие сорта, и применяется преимущественно на фабриках перевязочных средств для производства неиодоформенной марли и ваты.
Структура молекулы йодоформа представлена на рис. 1.
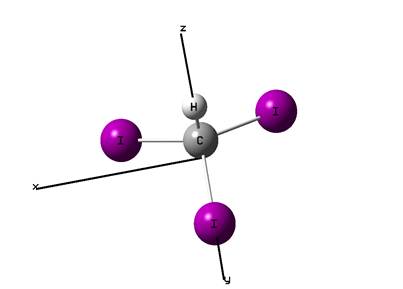
Рис. 1.1. Структура и пространственная ориентация молекулы йодоформа.
1.2. Применение хлороформа
Активное изучение обнаружило ряд уникальных свойств йодоформа. Под влиянием света и воздуха, тканевых выделений йодоформ медленно разлагается с выделением йода. Йод имеет антимикробное, дезорирующее, противовоспалительное и рассасывающее действие, способствует грануляции и очищению раны. На поверхности ран образуются альбуминаты йода, в результате проявляется вяжущее и анестезирующее действие, предотвращается раздражение рецепторов.
Таким образом, все эти свойства обуславливают широкое применение йодоформа в различных областях терапевтической и операционной медицины, сельского хозяйства и др.: лечение инфицированных и послеоперационных ран, язв, свищей, дерматитов, тендовагинитов, санитарной обработке скота перед убоем. Йодоформ применяют наружно в форме присыпки. Препарат наносят тонким слоем на инфицированные, послеоперационные раны, язвы или свищи с захватом здоровой ткани на расстояние 2–3 см. Повторные обработки проводят по хирургическим показаниям.
Для обработки свищей, ран с кармашками, а также для лечения ожогов бактериальной этиологии йодоформ растворяют в эфире. Для этого непосредственно перед применением готовят 10 % раствор препарата. В зависимости от величины ран и свищей количество раствора должно быть таким, чтобы полностью смочить обрабатываемую поверхность с учетом карманов и полостей ран.
При отитах раствор йодоформа заливают в ушную раковину животным, в зависимости от их размера в количестве от 1 до 5 мл. Обрабатывают обе ушные раковины, даже если в другой не выражены признаки заболевания. Обработку проводят однократно. Повторные обработки проводят по показаниям через 7 – 10 дней.
При лечении флегмон, лимфаденитов, дерматитов, тендовагинитов йодоформ смешивают с вазелином из расчета 5 – 10% йодоформа. Обработку проводят 1 – 2 раз в день в течение 5 –7 дней без перерыва.
После проведения санитарной обработки скота из-за устойчивого специфичного запаха препарата убой животных на мясо разрешается через 3 суток после обработки. При вынужденном убое животных ранее оказанного срока мясо используют на корм пушным зверям или перерабатывают на мясо-костную муку. Молоко дойных животных запрещается использовать для пищевых целей в течение 3-х суток после обработки. Оно может быть использовано после термической обработки в корм плотоядным животным.
1.3. Меры предосторожности при работе с йодоформом
Лица, работающие с йодоформом должны пользоваться спецодеждой. Во время работы запрещается курить, пить и принимать пищу. При попадании препарата на кожу или на слизистые оболочки его следует немедленно смыть обильным количеством проточной воды с мылом. При попадании внутрь – выпить несколько стаканов воды и вызвать рвоту, после чего выпить несколько стаканов воды с 2 – 3 таблетками активированного угля или другого адсорбента. Запрещается транспортировка и хранение йодоформа вместе с продуктами питания и фуражом, а также использование тары из-под него для бытовых целей.
По степени воздействия на организм при нанесении на кожу препарат относится к 3 классу умеренно опасных веществ по ГОСТ 12.1.007-76. Не обладает местно-раздражающим и резорбтивно-токсическим действием.
1.4.1. На основе карбоната натрия, йода и этилового спирта [1]
К раствору 20г кристаллического Na2CO3 в 100 мл воды прибавляют 10 г этилового спирта и нагревают на водяной бане до 700С. В нагретую смесь вносят при тщательном перемешивании небольшими порциями 10 г измельченного йода (раствор приобретает желто-бурую окраску, которая, однако, вскоре исчезает). После того, как внесен весь йод и жидкость обесцветилась, реакционную массу оставляют (под тягой) на 4-5 ч (можно и на ночь). Выпавший осадок отфильтровывают, многократно и тщательно промывают водой на фильтре и сушат на воздухе в темном месте. Выход около 2,5 г (30% от теоретического, считая на взятый в реакцию йод); т. пл. 1190С (после перекристаллизации из небольшого количества спирта).
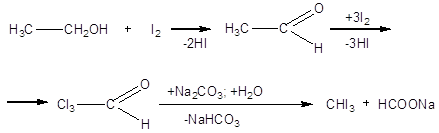
1.4.2. На основе ацетона, йода и щелочи [2]
Соединения типа ![]() или
или ![]() при обработке
свежеприготовленным раствором йода и едкой щелочи переходят в йодоформ, причем
промежуточно получаются трийодзамещенные продукты. Например:
при обработке
свежеприготовленным раствором йода и едкой щелочи переходят в йодоформ, причем
промежуточно получаются трийодзамещенные продукты. Например:
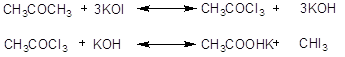
Это действие йода в щелочном растворе служит качественной реакцией на ацетон, этиловый спирт, ацетальдегид и т.д. и может служить также для количественного определения ацетона, спирта, молочной кислоты, окисляемых сначала до ацетальдегида (йодоформная реакция Либена).
1.4.3. Электролитический способ [3]
Получение йодоформа электролитическим путем. 50 частей йодистого калия растворяют в 300 частях воды и к этому раствору прибавляют около 30 частей 96%-ного спирта. Этот раствор подвергают электролизу при нагревании и непрерывном пропускании двуокиси углерода. Йодоформ выделяется в виде кристаллического порошка. Для получения йодоформа в форме больших кристаллов йодистый калий растворяют в 20%-ном спирте и подвергают электролизу, как указано выше.
Процесс протекает по следующему уравнению:
![]()
Действующим агентом здесь,
однако, является гипойодит щелочного металла: ![]()
В виде побочной реакции здесь идет главным образом образование йодата.
Для понижения скорости образования йодата и соответствующего значительного повышения выхода йодоформа прибавляют избыток йодида и особенно йодата.
1.4.4. На основе ацетона, йодистого калия, гипохлорита калия, этилового спирта [4]
Реактивы: ацетон 8г (10мл), йодистый калий 30г, гипохлорит натрия 25 г, этиловый спирт 300мл.
Аппаратура: колба круглодонная с механической мешалкой 1,5л; колба круглодонная 750мл; холодильник обратный, нагреватель для воронки, воронка Бюхнера, колба плоскодонная.
Методика: к раствору 30г (0,19моля) йодистого калия в 100 мл воды, помещенному в круглодонную колбу емк. 1,5л, снабженную мешалкой, приливают 10мл (8г-0,13моля) ацетона и, при перемешивании, небольшими порциями, около 300 мл 5%-ного водного раствора гипохлорита натрия. Конец реакции заметен по прекращению выделения желтого осадка йодоформа. Обычно он наступает после прибаления немного больше 300 мл раствора гипохлорита. Мешалку выключают, дают отстояться 0,5часа, отсасывают йодоформ на воронке Бюхнера и осадок тщательно промывают водой. Высушенный йодоформ перекристаллизовывают из этилового спирта (около 300 мл) и получают его в виде желтых кристаллов (т. пл. 1190) с характерным запахом. Выход – 17,5г (35%) теоретического.
Йодоформ получают действием на ацетон йодистого калия в пристутствии йода, действием на этиловый спирт, изопропиловый спирт или ацетон йодистого калия ил окислителей, таких, как гипохлориты и ли дихлорамины в щелочной среде. Электрохимический метод получения йодоформа заключается в электролизе раствора йодистого калия, содержащего карбонат натрия, в присутствии этилового спирта.
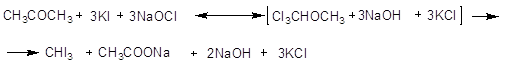
1.4.5. Промышленная методика производства йодоформа [5]
Молекулярный вес 394. Удельный вес 2,0. Лимонно-желтые гексагональные листочки или кристаллы в виде столбиков неприятного, сильно прилипчивого запаха. Растворимрим в 14000 ч. воды при 150С в 70 ч холодного винного спирта и в 10ч при 800, а также в эфире, хлороформе и сероуглероде. Выше 1200 разлагается. Получение соединяют с получением йодистого калия. Если йод, ацетон и едкое кали действуют друг на друга, то приблизительно 40% взятого йода превращаются в йодоформ, остаток дает йодистый калий и йодноватокислый калий. Сырьем для производства йодоформа служат:
Технический йод. Выбирают такие сорта, которые растворяются в едком кали с малым остатком и свободны от хлористого йода. На последнее обстоятельство нужно особенно обращать внимание в случае японского йода.
Ацетон. Берут такое количество, которое требуется для производства пороха.
Раствор едкого кали. Обычный продажный раствор технического едкого кали.
Спирт. Для некоторых сортов йодоформа для осаждения нужен спирт; он не участвует в процессе образования йодоформа, но служит только для получения определенных его сортов. Соответственно спиртовому законодательству различных стран его можно получать свободно, если он денатурирован метиловым спиртом или самим йодоформом.
2. Литературный обзор
2.1. Реакции нуклеофильного замещения галогеналканов
Благодаря доступности галогеналканов и легкости, с которой они вступают в реакции, круг этих реакций очень широк. Наиболее важные из них приведены в таблице 1.
Метилгалогениды CH3-X, первичные RCH2-X, вторичные R1R2CH-X, третичные R1R2R3-X алкилгалогениды взаимодействуют с нуклеофильными реагентами по разным механизмам в зависимости от строения алкила.
Таблица 1.
Реакции нуклеофильного замещения
|
Нуклеофил Nu |
Продукт реакции R-Nu |
|
НО- или Н2О R1O- или R1OH |
Спирт ROH Простой эфир ROR1 |
|
|
Сложный эфир |
|
NºC- |
Нитрил карбоновой кислоты R-CºN |
|
NO2- |
Нитросоединение R- NO2 |
|
NH3 |
Соль первичного амина RNH3+X- |
|
R1NH2, R1R2NH |
Соль вторичного или третичного амина RR1NH2+X-, RR1R2NH+ X- |
|
R1CºC- |
Алкины R1CºC-R |
|
R1C- |
R1C-R |
|
I- |
Иодиды R-J |
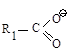
2.1.1. Бимолекулярное нуклеофильное замещение
Типичный механизм взаимодействия метилгалогенидов и первичных алкилгалогенидов с Nu - бимолекулярное нуклеофильное замещение SN2. По такому механизму протекает реакция бромметана с едким натром.
![]()
Стадии процесса. Нуклеофил атакует атом углерода с тыла, со стороны наиболее удаленной от брома (рис.2.1, а). Если сталкивающиеся частицы имеют достаточную энергию, то начинает образовываться связь углерод-кислород, а связь углерод-бром растягивается, атом углерода переходит в sp2-состояние. В этом состоянии атом углерода связан сразу с пятью атомами. Три атома водорода и углерод лежат в одной плоскости, а группы НО- и Br- располагаются на прямой, перпендикулярной этой плоскости (рис. 2.1, б). Отрицательный заряд на атоме кислорода уменьшился, так как кислород уже подал свою пару электронов на атом углерода, а отрицательный заряд на атоме брома увеличился, поскольку бром в определенной мере оттянул на себя пару электронов от углерода. Реакция заканчивается отщеплением иона брома и образованием ковалентной связи углерод-кислород, атом углерода опять становится тетраэдрическим (рис. 2.1, в).
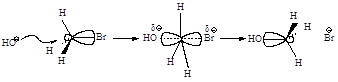
а б в
Рис.2.1. Бимолекулярное нуклеофильное замещение:а - исходные соединения: заряд локализован на атоме кислорода; б - переходное состояние (активированный комплекс), отрицательный заряд распределен между атомом кислорода и атомом брома; в - продукты реакции
Энергетическая диаграмма реакции (рис. 2.2) изображает изменение потенциальной энергии в ходе реакции нуклеофильного замещения.
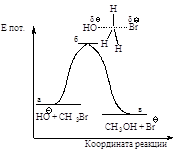
Рис.2.2. Диаграмма изменения потенциальной энергии в реакции бимолекулярного нуклеофильного замещения, SN2 - процесс согласованный одностадийный: а - энергия исходных веществ, б - энергия переходного состояния, в - энергия продуктов реакции.
Скорость реакции. Гидролиз бромистого метила является реакцией второго порядка, скорость его зависит от концентрации двух веществ и определяется по формуле V = K [R-Hal] [Nu]. Термин «бимолекулярное замещение» означает, что в скорость определяющей стадии участвуют две частицы. Поскольку разрыв связи углерод-уходящая группа и образование связи нуклеофил-углерод происходят одновременно, бимолекулярное нуклеофильное замещение называют согласованным процессом.
Концентрация нуклеофильного реагента. Высокая концентрация нуклеофила увеличивает скорость SN2- реакции.
Растворитель. Выбор растворителя диктуется следующими условиями:
а) достаточно хорошая растворимость реагентов,
б) лучшая сольватация переходного состояния по сравнению с исходными соединениями,
в) предотвращение побочных реакций.
Реакции, в которых из нейтральных молекул образуется полярное переходное состояние, значительно ускоряются при увеличении полярности растворителя: более полярный растворитель в большей степени стабилизирует полярное переходное состояние, чем исходную систему (рис.2.3.а).
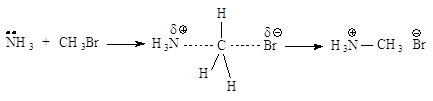
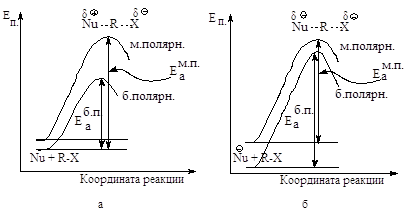
Рис.2.3. Влияние полярности растворителя на скорость SN2: а - повышение полярности растворителя стабилизирует АК в большей степени, чем исходное соединение, энергия активации уменьшается, скорость реакции увеличивается, б - повышение полярности растворителя стабилизирует исходную систему в большей степени, чем АК, энергия активации увеличивается, скорость реакции уменьшается.
Если в исходной системе имеется нуклеофил с полным отрицательным зарядом, то этот заряд стабилизируется в определенной степени в результате электростатического притяжения между молекулами полярного растворителя и ионом Nu- .
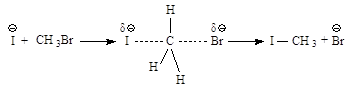
В активированном комплексе заряд распределен между атомом, образующим новую связь, и уходящей группой. Полярный растворитель будет стабилизировать и активированный комплекс и исходное состояние. Увеличение полярности растворителя несколько замедлит реакцию, так как замена менее полярного растворителя на более полярный увеличит в большей степени стабильность исходного соединения, в меньшей - активированного комплекса (рис. 2.3, б).
Наиболее подходящими для синтеза соединений и изучения механизма SN2 являются апротонные биполярные растворители, т.е. растворители с высокой диэлектрической проницаемостью, но не способные к образованию водородных* связей:
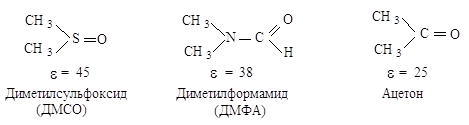
Апротонные растворители не могут сольватировать анионы за счет образования водородных связей с ними и химики называют их «голыми». Биполярные апротонные растворители особенно необходимы для осуществления реакций SN2 в случае применения малоактивных нуклеофилов. В реакции бромэтана (SN2) с гидроксиданионом лучшим растворителем является водный раствор этанола с массовой долей 80%; добавление воды к этанолу служит для предотвращения побочной реакции отщепления бромоводорода.
Уходящие группы. «Хорошими» уходящими группами являются те группы, которые, оторвавшись от атома углерода, образуют устойчивые анионы. Сильные основания являются обычно «плохими» уходящими группами, слабые основания - «хорошими» уходящими группами.
![]()
В этом ряду сила основания увеличивается, а способность быть хорошей уходящей группой уменьшается. Наилучшими уходящими группами являются ионы - сопряженные основания* сильных кислот, так как они являются очень слабыми основаниями (отрицательный заряд распределен).
*Водородная связь - связь между молекулами за счет электростатического притяжения между сильно протонированным атомом водорода одной молекулы и электроотрицательным атомом другой молекулы. Для образования водородной связи необходимо, чтобы электроотрицательными атомами были F, O, N.
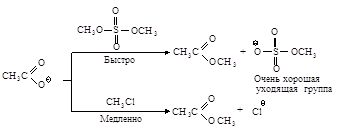
Cтереохимия. Бимолекулярное нуклеофильное замещение протекает с полным обращением конфигурации, т.е. происходит обращение каждой реагирующей молекулы.
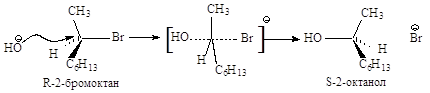
Конфигурация исходного 2-бромоктана при атаке хирального атома углерода с тыла меняется на противоположную, молекула субстрата выворачивается. Полное обращение конфигурации хирального углерода может служить доказательством SN2-механизма.
Обозначение конфигурации: Для этой цели наиболее широко используют символы R и S. Эта система обозначений предложена Р. Каном (Химическое общество, Лондон), К. Ингольдом (Университетский колледж, Лондон) и В. Прелогом (Федеральная высшая техническая школа, Цюрих).
Согласно этой системе, сначала определяют старшинство, или последовательность, заместителей, т. е. четырех атомов или групп, связанных с асимметрическим атомом углерода, исходя из правила старшинства.
Правило старшинства 1. Если с асимметрическим атомом углерода связаны четыре различных атома, то старшинство зависит от атомного номера, причем более старшим будет атом с большим атомным номером.
Если два атома являются изотопами одного элемента, то преимущество имеет атом с большим массовым числом. Например, в хлориодметансульфо- кислоте атомы, согласно их старшинству, располагаются в следующей последовательности: I > С1 > S > Н; в a-дейте-роэтилбромиде — Вг > С > О > Н.
*Всякое основание и кислота, между которыми существует соотношение называются сопряженными. Чем сильнее кислота, тем слабее сопряженное основание.
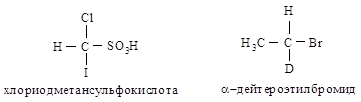
Правило старшинства 2. Если относительное старшинство групп нельзя определить с помощью правила 1, то необходимо провести аналогичное сравнение для следующих атомов в группах (и так далее, если необходимо, двигаясь дальше от асимметрического атома углерода). Иначе говоря, если асимметрический атом углерода связан с одинаковыми атомами, то следует сравнить заместители, связанные с каждым из этих первых атомов. Например, рассмотрим втор-бутилхлорид, в котором с асимметрическим атомом углерода связаны два углеродных атома. В СН3-группе следующими атомами являются Н, Н и Н; в С2Н5-группе – С, Н, Н.
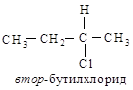
Поскольку углерод имеет больший атомный номер, чем водород, то С2Н5 старше. Таким образом, во втор-бутилхлориде заместители, согласно своему старшинству, располагаются следующим образом: С1 >С2Н5>СН3 > Н.
В З-хлор-2-метилпентане атомы С, С и Н изопропильной группы старше С, Н и Н этильной группы и полная последовательность заместителей будет следующей: С1 > изопропил > этил > Н.
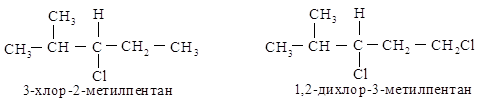
В 1,2-дихлор-З-метилпентане группа СН2Cl старше (С1, Н, Н) изопропильной (С, С, Н). Хлор имеет больший атомный номер, чем углерод, и то, что имеется два атома углерода и только один С1, не имеет значения. (один больший номер значит больше, чем два или три меньших.)
Правило старшинства 3. Атом, связанный двойной или тройной связью, считается соответственно за два или три атома. Таким образом,
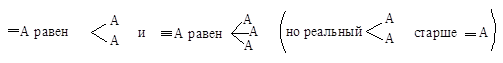
Например, в глицериновом альдегиде ОН-группа является старшей; СНО (О, О, Н) старше СН2ОН (О, Н, Н). Полная последовательность заместителей будет –ОН > –СНО > –СНаОН > –Н.
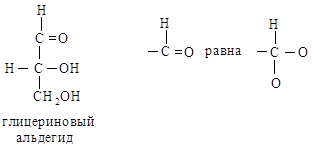
Фенильная группа С6Н5 рассматривается в виде одной из структур Кекуле:
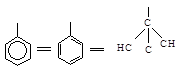
В 1-амино-2-метил-1-фенилпропане, например, фенильная группа (С, С, С) старше изопропильной (С, С, Н), но младше, чем N, который имеет больший атомный номер. Последовательность будет NН2 > С6Н5 > С3Н7 > Н.
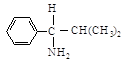
Обозначение конфигурации соединений с несколькими асимметрическими атомами: Существуют соединения с несколькими асимметрическими атомами Вопрос об их обозначении не представляет проблемы: обозначают конфигурацию каждого асимметрического атома углерода и, используя нумерацию, указывают, к какому атому относится каждое обозначение.
Рассмотрим, например, 2,3-дихлорпентаны. Каждый асимметрический атом углерода, С-2 и С-3, рассматривается по порядку без учета существования другого центра. Согласно правилам старшинства для С-2 получают последовательность С1 > СН3СН2СНСl > СНз > Н, а для С-3 — С1 > СН3СНС1> СН3СН2 > Н. (Почему СН3СНСl —«старше», чем СН3СН2—?)
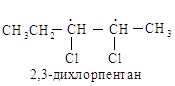
При рассмотрении модели определенного стереоизомера сначала концентрируют внимание на С-2 (игнорируя С-3), а затем на С-3 (игнорируя С-2). Стереоизомер I обозначают как (2S, 3S)-2,3-дихлорпентан, а другие изомеры — как (2R, ЗR), (2S, ЗR) и (2R, 3S).
Рассмотрим таким же образом 2,3-дихлорбутан. В этом случае два асимметрических aтома углерода эквивалентны, и нет необходимости их нумеровать.
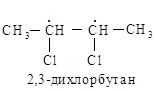
Например, в случае СНСlВгI с асимметрическим атомом углерода связаны четыре различных атома, и старшинство их зависит только от атомного номера, причем, чем больше атомный номер, тем старше заместитель. Таким образом, в порядке уменьшения их старшинства атомы располагаются в следующем порядке: I > Вг > С1 > Н.
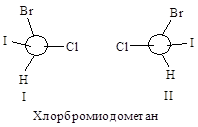
Затем молекулу располагают, так, чтобы младшая группа была направлена от наблюдателя, и рассматривают расположение оставшихся групп. Если старшинство этих групп уменьшается по часовой стрелке, то конфигурацию обозначают символом R (от латинского rectus — правый); если же старшинство этих групп уменьшается против часовой стрелки, то конфигурацию обозначают символом S (от латинского sinister — левый).
Таким образом, конфигурации I и II выглядят следующим образом:
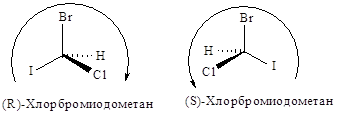
и обозначаются соответственно символами R и S.
Полное название оптически активного соединения отражает и конфигурацию и направление вращения, как, например, (S)-(+)-втор-бутилхлорид. Рацемическую модификацию можно обозначить символом R,S, например (R,S)-втор-бутилхлорид.
Конечно, нельзя путать направление оптического вращения соединения (такого же физического свойства реального вещества, как температура кипения или плавления) с направлением нашего взгляда, когда мы мысленно располагаем молекулу каким-то определенным условным образом. Пока для определенного соединения экспериментально не установлена связь между конфигурацией и знаком вращения, нельзя сказать, знак (+) или (–) соответствует (R)- или (S)-конфигурации.
Сила нуклеофила. Нуклеофил - частица, имеющая пару электронов, которую она может отдать атому углерода, несущему целый или частичный положительный заряд. Чем сильнее атом удерживает пару электронов, тем меньше его способность выступать в качестве нуклеофила.
Активность аниона выше, чем активность нейтральной молекулы.
![]()
Нуклеофильный реагент, предоставляющий пару электронов электронодефицитному атому углерода, способен подавать эту пару атому водорода и отщеплять его, превращаясь в кислоту, т.е. каждый нуклеофильный реагент является основанием. Нуклеофильная реакционная способность и основность изменяются параллельно у реагентов, в которых пара электронов находится на одном и том же атоме или неподеленная пара электронов находится у атомов элементов, принадлежащих одному периоду.
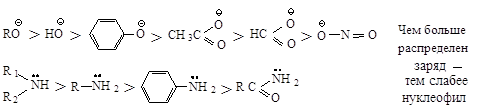
![]()
Реакционная способность нуклеофилов с атомами, находящимися в одной группе, зависит от положения элемента в этой группе: чем больше электроотрицательность атакующего атома, тем более реакционноспособен нуклеофил.
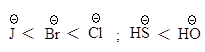
В протонных растворителях нуклеофильность аниона тем выше, чем больше размер иона.
![]()
Такой порядок изменения нуклеофильности в протонном растворителе объясняется тем, что анионы разного размера в протонном растворителе в различной степени сольватированы за счет образования водородных связей: анион малого размера с концентрированным зарядом сольватируется сильнее и стабилизирован в большей степени, чем анион большего размера, в котором отрицательный заряд распределен в большей степени.
Сила нуклеофила играет важную роль: например, неопентилбромид реагирует с этилат-ионом по SN2-механизму, а с этиловым спиртом по SN1 -механизму. Сильный нуклеофил (сильное основание), этилат-ион, выталкивает ион галогена из молекулы, тогда как слабый нуклеофил, этиловый спирт, ждет, пока ион галогена отойдет.
Реакционная способность. При рассмотрении реакционной способности галогеналканов в реакциях нуклеофильного замещения следует изучать влияние двух факторов: пространственного (стерического) и электронного. В случае бимолекулярного нуклеофильного замещения наиболее важную роль играет стерический фактор. По мере увеличения числа и объема заместителей у атома углерода - реакционного центра возможность достижения активированного комплекса уменьшается. Это могут быть как алифатические, так и ароматические заместители или те и другие.
В SN2-реакциях реакционная способность уменьшается в ряду:
СН3–Х > RСН2–Х > R1R2СН–Х > R1R2R3С–Х

2.1.2. Мономолекулярное нуклеофильное замещение
Третичные алкилгалогениды реагируют по механизму SN1 (мономолекулярное нуклеофильное замещение).
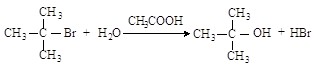
Cтадии процесса. Реакция протекает в две стадии. Первая стадия - гетеролитический разрыв связи углерод-галоген - медленная.
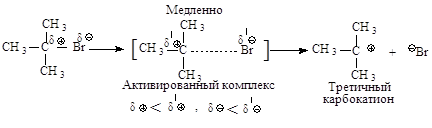
Вторая стадия - образовавшийся карбокатион практически мгновенно взаимодействует с нуклеофилом молекулой воды.
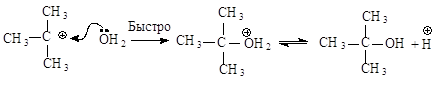
В отличие от механизма SN2 разрыв связи С-Х и образование новой связи С-Nu протекает не одновременно, а последовательно.
Энергетическая диаграмма реакции изображает изменение потенциальной энергии в ходе двух стадийного мономолекулярного замещения.
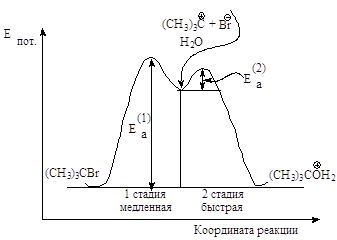
Рис.2.4. График изменения потенциальной энергии в ходе мономолекулярного нуклеофильного замещения. SN1-двухстадийный последовательный процесс.
Скорость реакции. В медленной стадии, определяющей скорость реакции, принимает участие только одна молекула, поэтому механизм называют мономолекулярным замещением. Скорость реакции зависит от концентрации галогеналкана и определяется по формуле V = K[R-Hal].
Концентрация нуклеофильного реагента. Низкая концентрация нуклеофила способствует SN1-реакции.
Перегруппировка. В медленной стадии реакции образуется карбокатион - частица, способная к перегруппировке. Галогеналканы могут реагировать по механизму SN1 через стадию перегруппировки первоначально образующегося карбокатиона: если в результате 1,2-сдвига отрицательно заряженной частицы образуется более устойчивый карбокатион, то происходит перегруппировка.
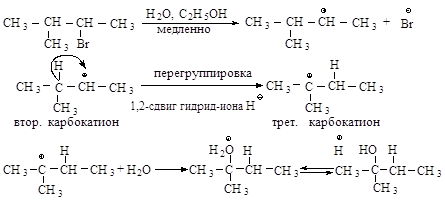
Перегруппировка считается признаком SN1-механизма.
Растворитель. Переходное состояние SN1-реакции более полярно, чем исходное состояние. Увеличение полярности растворителя способствует большей сольватации переходного состояния по сравнению с сольватацией исходного соединения. Это приводит к росту скорости реакции.
При проведении SN1-реакции используют протонные растворители с высокой диэлектрической проницаемостью, способные образовывать водородные связи.
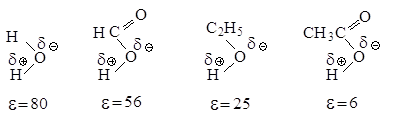
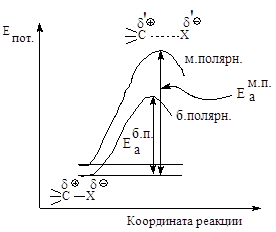
Рис.2.5. Влияние полярности растворителя на скорость реакции мономолекулярного нуклеофильного замещения
В растворителе, имеющем высокую диэлектрическую проницаемость, легче протекает ионизация галогеналкана, но, в отличие от апротонных растворителей, в протонном сольватируется не только карбокатион, но и ион галогена, образуя с ним водородные связи.
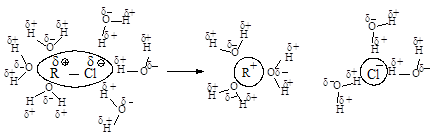
Сольватация сопровождается выделением значительного количества энергии, которая может быть затрачена на ионизацию новых исходных молекул, что приводит к ускорению реакции.
Сольватация нуклеофила не влияет на скорость реакции, так как даже стабилизированный сольватацией нуклеофил быстро реагирует с карбокатионом.
Стереохимия. В карбокатионе, образующемся в медленной стадии, связи sp2 -гибридизованного атома углерода расположены в одной плоскости. Если SN1-замещение протекает у хирального атома углерода, то образующийся плоский карбокатион становится ахиральным.
Последующая атака нуклеофильного реагента происходит с одинаковой вероятностью и с той и с другой стороны плоского карбокатиона. Следовательно, половина образующихся молекул будет иметь ту же конфигурацию, что и исходное соединение, а половина - будет его зеркальным изображением, т.е. образуется эквимолекулярная смесь энантиомеров - рацемат. Такая реакция приводит к оптически неактивному продукту.
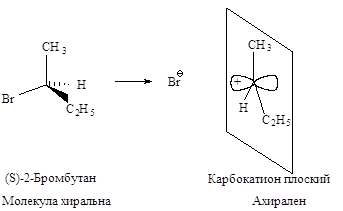

В том случае, если уходящий галогенид-ион не успевает отойти от реакционного центра, он затрудняет атаку нуклеофила со своей стороны. Это приводит к образованию большего количества изомера с конфигурацией, противоположной конфигурации исходного соединения. Тогда имеет место частичная рацемизация.
Реакционная способность. Главным фактором, определяющим реакционную способность в SN1-реакции, является электронный фактор - устойчивость образующегося в медленной стадии реакции карбокатиона. Чем устойчивее карбокатион, тем легче он образуется, тем быстрее протекает замещение.
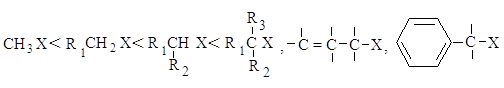
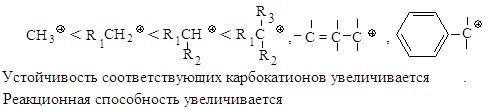
2.1.3. Сравнение реакций SN 1 и SN 2
Каждый из этих механизмов в чистом виде встречается очень редко. Реакционная способность в SN2-реакции уменьшается при переходе от СН3-Х к первичным RCH2-X, для вторичных - она гораздо меньше и появляется значительный вклад SN1-реакций. При переходе от вторичных к третичным галогеналканам реакционная способность в SN1-реакции резко возрастает.
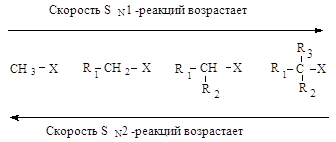
Изменяя условия протекания реакций, можно направить процесс преимущественно по тому или иному механизму.
Таблица 2.2
Влияние условий реакции на относительное значение двух механизмов
| Механизм | Растворитель | Сила нуклеофила |
Концентрация нуклеофила |
|
SN1 |
Протонный | Более слабый | Меньшая |
|
SN2 |
Апротонный | Более сильный | Бóльшая |
2.2. Реакции отщепления (элиминирование)
В ходе реакции элиминирования от молекулы галогеналкана отщепляются два фрагмента: ион галогена (уходящая группа) от Сa и протон - от соседнего атома углерода Сb. Такой тип реакций называется b-элиминированием.
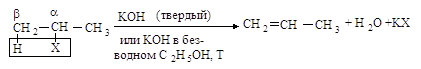
2.2.1. Бимолекулярное отщепление Е2
Реакция отщепления галогеноводорода от первичных галогеналканов протекает по бимолекулярному механизму Е2.
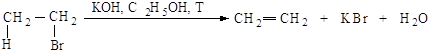
Механизм Е2. Основание НО– атакует водород (рис.2.6.а) при Сb, подает ему пару электронов и начинает образовывать с ним связь, связь Сb-Н ослабевает, пара электронов, связывавшая углерод и водород, освобождается и атакует атом галогена с тыла., одновременно растягивается связь Сa -Br (процесс согласованный). Возникает активированный комплекс: атомы углерода Сa и Сb переходят из sp3-гибридного состояния в состояние, близкое к sp2-состоянию, освобождающаяся пара электронов затрачивается на образование p-связи (рис.2.6.б) . Затем связи углерод- водород и углерод-галоген разрываются, атомы углерода переходят в sp2-состояние, образуется p-связь (рис.2.6.в).
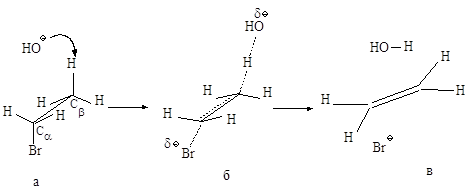
Рис.2.6. Механизм реакции бимолекулярного элиминирования; а – антикомпланарное расположение связей Сa—Br и Сb—H, б – переходное состояние, в конечные продукты.
Энергетическая диаграмма реакции изображена на следующем графике:
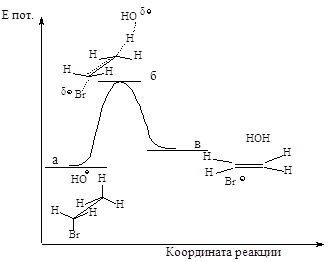
Рис.2.7. График изменения потенциальной энергии в ходе Е2.
Связи, разрывающиеся в активированном комплексе, должны быть в транс-положении друг к другу. Такая геометрия позволяет электронной паре, освобождающейся при связывании протона с основанием, атаковать с тыла атом углерода Сa и вытеснять галоген. Копланарность двух связей субстрата: Сa -Br и Сb -Н, которые разрываются в активированном комплексе, обеспечивает максимальное перекрывание р-орбиталей, то есть способствует процессу образования p-связи. Такая комбинация условий называется стереоэлектронным требованием.
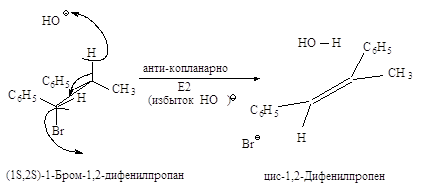
Ниже эта реакция изображена в проекциях Ньюмена.
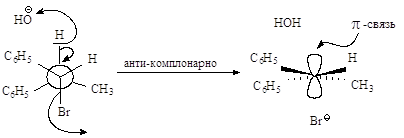
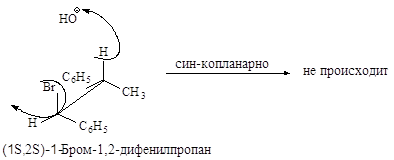
Реакция, в результате которой из данного стереоизомерного соединения получается в качестве продукта только один стереоизомер, называется стереоспецифической реакцией.
Cкорость реакции зависит от концентрации галогеналкана и основания и определяется по формуле V= K[R-Br] [O–].
Направление элиминирования - правило Зайцева: основным продуктом реакции отщепления от галогеналканов с двумя не эквивалентными Сb является наиболее устойчивый (наиболее алкилированный) алкен.
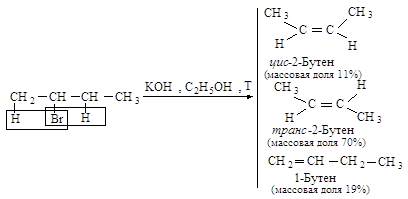
Основным продуктом реакции элиминирования 2-хлорбутана является транс-2-бутен (соотношение цис- и транс-изомеров 1:6). Активированный комплекс, возникающий при образовании транс-изомера, менее пространственно затруднен и более устойчив, и, следовательно, скорее образуется, чем активированный комплекс, ведущий к цис-изомеру.
Реакционная способность галогеналканов в реакциях Е2 изменяется в ряду:
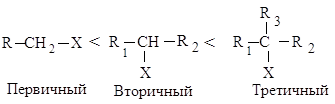
Такое увеличение реакционной способности галогеналканов при переходе от первичных к третичным обусловлено увеличением устойчивости образующихся алкенов.
2.2.2. Мономолекулярное отщепление Е1
Третичные галогеналканы реагируют по мономолекулярному механизму.
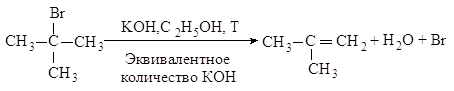
Механизм. Реакция протекает в две последовательные стадии. Первая стадия аналогична первой стадии мономолекулярного нуклеофильного замещения:
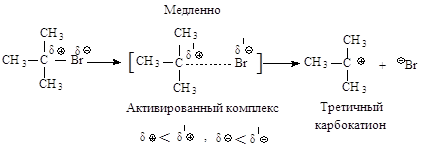
Во второй стадии основание атакует водород при Сb - атоме.
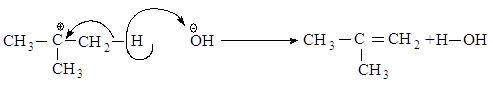
Реакционная способность галогеналканов увеличивается при переходе от первичных к третичным. Это обусловлено увеличением устойчивости карбокатионов, образующихся в медленной стадии.
2.2.3. Сравнение реакций нуклеофильного замещения и элиминирования
В реакциях замещения донор электронной пары отдает свою пару электронов атому углерода Сa, связанному с галогеном - в этом случае он является нуклеофилом. Тот же донор электронов может отдавать свою электронную пару атому водорода, связанному с Сb - в этом случае он является основанием в реакциях элиминирования.
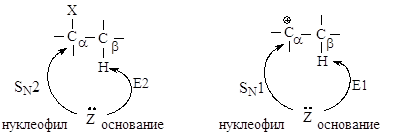
Реакции элиминирования и нуклеофильного замещения в этом случае являются конкурирующими. При переходе от первичных ко вторичным и далее третичным галогеналканам элиминирование происходит все в большей степени.
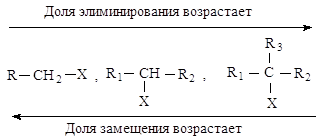
Cильные основания в большей степени способствуют элиминированию. Выход продуктов элиминирования увеличивается за счет продуктов замещения также с повышением температуры.
2.3. Методы синтеза галогеналканов
2.3.1. Галогенирование алканов
2.3.2. Присоединение галогенводородов к олефинам
2.3.3. Замещение гидроксильной группы спиртов на галоген
· действием галогенводородов:
![]()
или
![]()
· галогенидов фосфора:
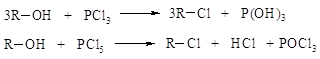
· или хлористым тионилом:
![]()
Список литературы
1. Агрономов А.Е., Шабаров Ю.С. Лабораторные работы в органическом практикуме. Изд. 2-е, М., Химия, 1974, с. 83
2. Губен И. Методы органической химии. Том III, выпуск 3, М., ОНТИ, 1935,437
3. Кацнельсон М. М. Приготовление синтетических химико-фармацевти-ческих препаратов. Л., Госхимиздат, 1933,132.
4. Препаративная органическая химия. Изд. 2-е, М., Гохимиздат, 1964,188.
5. Швицер Ю. Производство химико-фармацевтических и технохимичес-ких препаратов. М.-Л., ОНТИ, 1934, 106.
6. Шабаров Ю.С. Органическая химия: В 2-х кн. - М.: Химия, 1994.- 848 с.
7. ПетровА.А., Бальян Х.В., Трощенко А.Т. Органическая химия. - М.: Высш. шк., 1973. - 623 с.
8. Моррисон Р., Бойд. Органическая химия. - М.: Мир, 1974. - 1132 с.
9. Терней А. Современная органическая химия: В 2 т. - М.: Мир, 1981. - Т.1 - 670 с; Т.2 - 615 с.
10. Робертс Дж., Кассерио М. Основы органической химии: В 2 т. - 2-е изд. -М.: Мир, 1978. - Т.1 - 842 с; Т.2 - 888 с.
11. Веселовская Т.К., Мачинская И.В., Пржиялговская Н.М. Вопросы и задачи по органической химии. - М.: Высш. шк., 1977. - 230 с.
12. В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, - 708 с.
© 2009 База Рефератов