
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Реферат: Окисление парафиновых углеводородов
Реферат: Окисление парафиновых углеводородов
Окисление парафиновых углеводородов
Ряд процессов жидкофазного окисления углеводородов в настоящее время реализованы как крупнотоннажный производства, например СЖК [2], высших жирных спиртов и др. [3].
Изучения жидкофазного окисление насыщенных углеводородов оказалось весьма плодотворным для установления общих закономерностей процесса окисления. На примере окисления индивидуальных углеводородов и их смесей получены фундаментальные знания о механизме радикальных реакций [3,4-7].
Некоторые особенности процесса жидкофазного окисления парафиновых углеводородов.
Окисления парафиновых углеводородов хорошо изученный процесс [7,8,10-12].
Известно, что окисление парафиновых углеводородов молекулярным кислородом приводит к образованию большого число промежуточных и конечных кислородсодержащих продуктов: перекисей, спиртов, карбонильных соединений, кислот, эфиров, а также бифункциональных соединений.
Найден ряд катализаторов процесса окисления углеводородов, таких как, растворимые комплексы титана [9], хлорид платины [14], комплексы ванадия (5V) [15], Pd, Pt, Co, Fe нанесенные на носитель, например, на цеолита [16], система на основе Ti, Zr, V, Cr, Mo, W, Mn, Fe и имида [19], система из растворимых соединений кобальта и хрома [21], мультиоксиды металлов [23], алкилперокси- комплексы трехвалентного кобальта [25], смесь азотной кислоты и уксусного ангидрида [26], комплексы марганца и органических кислот содержащих ароматических фрагментов [29], комплексы металлов [30], SiO2 , AI2O3 , ZrO и другие на носителе [31] комплексы металлов, содержащую имидную группировку [32], система на основе Bi, V, Mo, Ag [33], Мn содержащий катализатор, нанесенный на молекулярный сита [34]. Известны каталитические системы ведущие процесс окислению углеводородов селективно [13,18,22,24,28].
К настоящему времени считается доказанным, что в случае окисления предельных углеводородов гидроперекиси единственные первичные промежуточные продукты.
Изучения строения образующихся при окислении гидроперекисей показало, что строение углеводородного радикала R в гидроперекиси R'OOH сохраняется таким же, как и в исходном углеводороде RH [3].
Образующиеся при окислении радикалы R'02 взаимодействует с молекулой исходного углеводорода, отрывая атом водорода и образуя гидроперекиси по реакции
![]()
При окислении разветвленных парафинов с двумя третичными связями С - Н в большом количестве были обнаружены дигидроперекиси. Окисление проводили при 115 — 120°С до глубины 5 -8 % (мол.) [3]. В начальный период окисления свободные радикалы образуются при взаимодействии исходного углеводорода с растворенным в нем кислородом
![]()
Радикал R* присоединят к себе молекулу кислорода и превращается в перекисный радикал RO2•, который далее отрывает атом водорода от молекулы углеводорода и образует гидроперекись и свободный радикал R•, продолжающий цепь. В процессе окисления накапливается гидроперекись, молекулы который сравнительно медленно распадаются на радикалы, например по реакции
![]()
Это приводит к увеличению скорости образования свободных радикалов. Процесс распада промежуточных гидроперекисей на радикалы представляет собой реакцию вырожденного разветвления цепей [3].
В целом механизм цепного окисления углеводородов может быть представлен следующим образом [41]:
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Имеющийся в настоящее время экспериментальный материал подтверждает цепную схему окисления углеводородов.
Чем выше скорость образования свободных радикалов, тем выше их концентрация, тем чаще происходит встреча и рекомбинация (или диспропорционирование) двух свободных радикалов и тем короче цепь обрыв цепей может происходит при взаимодействии свободного радикала со стенкой реактора (обрыв на стенке), а также по бимолекулярной реакции между двумя свободными радикалами (квадратичный обрыв). В жидкой фазе диффузия свободных радикалов у стенке весьма затруднена из - за высокой вязкости среды. По этому в цепных жидкофазных реакциях осуществляется квадратичный обрыв цепей по реакциям:
![]()
![]()
![]()
где: МП - молекулярные продукты.
Эти реакции протекают с малой энергией активации, в 4,1 - 8,4 кДж/моль.
Реакции между двумя вторичными перекисными радикалами приводит к образованию спирта и кетона.
Происходящее в процессе окисления превращение молекулы углеводорода последовательно в гидроперекись, спирт и кетон сохраняет исходный углеводородный скелет молекулы.
В процессе окисление кислоты декарбоксилируются сравнительно медленно, и их состав практически не меняется в ходе окисления. Среди кислот, образующихся при окислении н- декана обнаружены окси- и кетокислоты (15- 18% от общего число кислот). Однако эти кислоты образуются не из жирных кислот, а параллельно с ними.
Скорость образования уксусной кислоты составляет только 30% от скорости окисления кетона. Следовательно, механизм разрыва а - С - С- связи в окисляющихся парафинах не единственный, и по этому направлению образуются меньше половины низших кислот при окислении парафинов.
При большой скорости растворения кислорода его концентрация в окисляющемся веществе близка к насыщению; процесс протекает в кинетической области, т.е. не зависит от скорости растворения и диффузии кислорода в жидкой фазе. При очень быстром окислении диффузия кислорода в жидкость может оказаться лимитирующий стадией процесса окисления. В этом случае реакция будет протекать в диффузионной области. Поэтому, при изучении закономерности реакции окисления протекает в кинетической области [3].
Так как для подавляющего большинство органических соединений, окисляющихся в жидкой фазе, энергия разрыва связи С - Н меньше 377 кДж/моль, то в жидкой фазе зарождение цепей должно происходить преимущественно по тримолекулярной реакции, что доказано Е.Т. Денисовым [7].
Перекисные радикалы в среде окисляющегося углеводорода могут не только взаимодействовать с компонентами реагирующей смеси (например, с исходным углеводородом), образуя гидроперекиси, но и подвергаться распаду с образованием стабильного продукта и нового свободного радикала, как это наблюдается при окислении углеводородов в газовой фазе[3].
Н.С.Ениколопяном показано, что в сложных цепных реакциях, протекающих с образованием ряда стабильных промежуточных продуктов, длина цепи может меняться по ходу реакции, что в свою очередь приведет к изменению скорости реакции, остановка окисления углеводородов задолго до полного расходования исходных веществ, постоянная скорость протекания реакции до очень больших глубин превращения (наблюдаемая для метана, бензола и др.) , несовпадение порядка реакции, определённого по ходу процесса, с определённым по начальной концентрации исходного углеводорода, автокатализ промежуточными и конечными продуктами, катализирующее и ингибирующее действие одних и тех же веществ в различных реакциях могут получить удовлетворительное объяснение в рамках представлений о том, что если в результате реакции стабильных промежуточных продуктов реакции с радикалом образуется радикал, более активный, чем исходный, то имеет место удлинение цепи. В противном случае по мере накопления стабильных промежуточных продуктов длина цепи уменьшается [39] .
В условиях окисления гидроперекиси могут расходоваться не только при взаимодействии со свободными радикалами и по реакции разветвления, обычно протекающей медленно, но и другими путями, которые для общности называют не цепным расходованием. В некоторых реакциях окисления такой не цепной путь распада оказывается доминирующим. Так, при окислении альдегидов образующаяся над кислота реагирует с исходным альдегидом с образованием кислоты.
В присутствии кислот гидроперекиси подвергаются гетеролитическому расщеплению, что приводит к автоторможению в реакции окисления.
Работами Н.М.Эмануэля [3] показано, что ряд реакций окисления углеводородов прекращаются задолго до полного израсходования исходного вещества.
Вопросы автоторможения реакций окисления подробно изучены Е.Т.Денисовым [40]. Было показано, что в начальный период окисление углеводорода осуществляется за счет взаимодействия RH с перекисными радикалами:
![]()
И скорость реакции определяется концентрацией перекисных радикалов. По мере накопления продуктов окисления - гидроперекисей, спиртов, кетонов, кислот - перекисные радикалы вступают в реакцию взаимодействия с этими продуктами.
При цепном распаде вторичной гидроперекиси радикал R02 заменяется на свободный гидроксил: Реакция RO2• со спиртом приводит к образованию оксигидроперекисного радикала: Реакция с кислотами приводит к выделению СО2 и другому R1O2• радикалу:
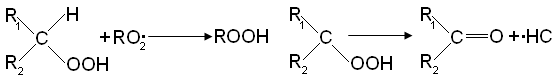
Реакция RO2• со спиртом приводит к образованию оксигидроперекисного радикала:

Реакции с кислотами приводит к выделению СО2 и другому R1O2• радикалу:

В развившейся реакции исходный углеводород может взаимодействовать с различными свободными радикалами, и скорость его окисления зависит не только от общей концентрации радикалов, но и от их состава. В реальных процессах окисления скорость реакции по ходу изменяется не только в зависимости от скорости вырожденного разветвления цепей , но и в зависимости от состава радикалов.
Специальными исследованиями и кинетическими расчетами было установлена [38], что в сложных цепных реакциях, протекающих с образованием молекулярных промежуточных продуктов, состав радикалов неизбежно меняется в ходе реакции вследствие изменения состава продуктов. Изменяющийся состав радикалов воздействует не только на суммарную активность радикалов, но и на их общую концентрацию путем изменения скорости квадратного обрыва цепей. Таким образом, механизм воздействия состава радикалов на скорость сложной цепной реакции таков, что ускорения реакции всегда ограничено, а её замедления может быть сколь угодно сильным. Это обстоятельство и является фундаментальной причиной широко распространенного явления самоторможения реакций окисления.
Продукты распада радикала R02 были обнаружены при жидкофазном окислении н. бутана, изопропилбензола и циклогексана в металлических реакторах.
Интересным представляется наблюдение [3]. О том, что в металлических реакторах продукты, образующейся с разрывом углеродного скелета в случае окисления бутана, составляют около 10-12% от прореагировавшего бутана, тогда как при окислении н. декане было обнаружено ни карбонильных соединений, ни спиртов, содержащих в своей молекуле меньшее число атомов углерода, чем исходный декан.
Это на первый взгляд кажется удивительным, поскольку оба углеводорода принадлежат к одному и тому же классу- к парафинам нормального строения. В действительности никакого различия механизмом окисления н. декана и н. бутана не существует, а наблюдаемое расхождение в составе продуктов этих реакций объясняется, влиянием нержавеющей стали на распад радикала R-2 при проведении процесса в металлических реакторах.
Под влиянием металла происходит также уменьшения периода индукции и увеличение суммарной скорости окисления, определяемой по скорости расходования бутана. При этом скорости накопления продуктов распада увеличиваются в несколько раз больше, чем скорости накопления продуктов гидроперекисного направления. Ускорение реакции связано, по-видимому, с увеличением скорости разветвления цепи за счет увеличения скорости распада гидроперекиси под действием металлической поверхности.
Реакция распада радикала RO2• предшествует его изомеризация с переходом свободной валентности от атома кислорода к одному из соседних атомов в радикале. Изомеризация перекисного радикала происходит наиболее легко в случае, когда в третичные атомы углерода находятся в β - положении относительно друг - друга.
При этом если свободная валентность, переходить к атому углерода, то вслед за изомеризацией радикала происходит разрыв связи С-С. Состав продуктов окисления сжиженного бутана при температуре 145°С и давлении 50 атм. В реакторе из нержавеющей стали отличается от состава продуктов окисления н. бутана в стеклянном реакторе. Наряду с соединениями, образующимися из гидроперекиси (метил этил кетон, вторичный бутиловый спирт, уксусная кислота), обнаруживается существенным количества веществ, содержащих меньшее число атомов углерода, чем исходный бутан (ацетальдегид, ацетон, метиловый и этиловый спирт и другие). Показано, что эти соединения не является продуктами дальнейшего превращения гидроперекиси, так как при термическом разложении гидроперекиси в атмосфере азота в тех же условиях, в которых проводится процесс окисления бутана, образуются только бутиловый спирт и метил этил кетон.
Если изомеризация
радикала ![]() сопровождается
переходом свободной валентности к атому водорода, а не к углеводородному атому,
то распад радикала происходит с разрывом связи С – Н . так как в этом случае
углеродный скелет не разрушается, то образуются продукты, практически
неотличимы от соединений, получающихся в результате превращений гидроперекиси.
сопровождается
переходом свободной валентности к атому водорода, а не к углеводородному атому,
то распад радикала происходит с разрывом связи С – Н . так как в этом случае
углеродный скелет не разрушается, то образуются продукты, практически
неотличимы от соединений, получающихся в результате превращений гидроперекиси.
В реакциях окисления углеводородов гидроперекиси очень часто главные, но не единственные первичные молекулярные продукты окисления. Во многих случаях параллельно с гидроперекисями образуются циклические и полимерные перекиси, окиси и другие продукты окисления.
Таким образом, из литературы известно, что металлы, контактирующие с окисляющимся углеводородом не всегда инертны к процессу окисления.
Катализ процесса окисления солями металлов переменной валентности.
При окислении углеводородов в качестве катализаторов обычно применяют органические соли кобальта, марганца, железа, меди, хрома, свинца, никеля. Перманганат калия, например, служит катализатором окисления парафина кислородом воздуха в производстве жирных кислот [2]. Катализаторы позволяют, проводит окисления при более низкой температуре, т.е. в более мягких условиях и таким образом уменьшают количество нежелательных продуктов глубокого окисления [3].
В реакциях окисления углеводородов механизм соленого катализа очень сложный. Ускоряя реакцию окисления, катализатор испытывает обратное воздействие продуктов окисления, что приводит к протеканию процесса в несколько последовательных стадиях.
Каталитические действие соединений металлов переменной валентности указывает на цепной характер окисления.
Изучение особенностей жидкофазного окисления углеводородов инициированного солями металлов, проведенное В.Г.Фрейдиным [80],показало, что период индукции при использовании двухвалентных металлов (Мn) значительно длиннее, чем при применении трехвалентных (Сr); период индукции увеличивается (в изученных пределах) с повышением содержания двухвалентного металла; спектры поглощения образующихся в индукционном периоде соединений металлов в высшем валентном состоянии соответствуют спектрами поглощения известных комплексных органических солей этих металлов; анализ стеарата кобальта, изменившегося и индукционном периоде окисления керосина; дает возможность приписать ему строение частично гидролизованного многоядерного комплексного соединения, присутствие спиртов ускоряет переход металлов в высшее валентное состояние [85]. Известно, что в зависимости от валентного состояния, ионы металлов переменной валентности могут присоединять или отдавать один электрон какой-либо валентно насыщенной молекуле. Это неизбежно приводит к образованию свободных радикалов, ускоряющих цепной процесс окисления.
![]()
![]()
Ион трех валентного металла в среде реакционной массы образует многоядерный катион:
![]()
Реакция с участием многоядерного катиона ускоряют реакцию и приводят к образованию продуктов окисления:
![]()
![]()
где: Ас- - анион кислоты (продукта окисления).
Таким образом, в начале процесса окисления с участием двухвалентных ионов металлов переменной валентности замедление реакции объясняется обязательной последовательностью процессов:
первичного инициирования, необходимая продолжительность которого увеличивается в результате большой потребности в первичных продуктов окисления (гидроперекисей), участвующих в образовании комплекса;
реакции образования комплекса;
процесса разрушения комплекса с образованием ионов и радикалов осколков комплекса, инициирующих развитую реакцию [31].
Было показано [4], что под действием кислорода эполеты металлов разлагаются, образуя две молекулы кислоты. Для практического использования катализатора большое значение имеет вопрос о стабильности жирных кислот в условиях технологического режима окисления. Тем не менее роль катализатора в процессе окисления высокомолекулярных жирных кислот выяснена недостаточно. Была изучена окисляемость фракций синтетических жирных кислот Сю -Ci3 и Си - С20. при переменном температурном режиме и в присутствии 0,2% КМпОд кислоты Сю - Сю окисляются незначительно, а кислоты Сю - Сго с большими скоростями. Кислотное число водорастворимых кислот по мере протекания каталитического окисления непрерывно повышается. Это свидетельствует о том, что кислоты обогащаются низкомолекулярными веществами. Наиболее эффективно процесс окисления ускоряется некоторой оптимальной концентрацией Мn, ровной -0,1%. Избыток КМnО4 по сравнению с оптимальной концентрацией или увеличение доли щелочного металла в составе катализатора приводят к разному уменьшению скорости процесса, в то время как один марганец влияет на скорость окисления гораздо слабее, чем в смеси с калием. Таким образом, основные ингибирующие функции в данном случае принадлежат, по-видимому, соединением щелочного металла [3].
Воздействие катализатора на реакцию окисления проявляется тем отчетливее, чем ниже температура окисления. При невысокой температуре катализированной окисления намного быстрее некатализированного. С повышением температуры различие в скоростях уменьшается. Это связано с тем, что предостаточно высокой температуре цепной процесс окисления способен к быстрому развитию в отсутствие катализатора, а солей катализатора выпадает в осадок на сравнительно неглубоких стадиях процесса вследствие накопления кислот и почти не участвует в реакции [4].
В промышленном производстве синтетических жирных кислот окисляют смесь (1:2) исходного парафина с возвратным, т.е. полученным после отделения продуктов реакции. Необходимая условия нормального протекания процесса присутствие катализатора. Обычно используют окиси марганца, содержащие щелочь или перманганат калия в количестве 0,08-0,1% от веса загрузки, считая на марганец. Реакция проходит при переменном температурном режиме 125-105°С. Постепенное снижение температуры по мере накопления продуктов окисления предотвращает обогащение жирных кислот побочными веществами, уменьшает концентрацию полифункциональных соединений окси кислот и т.п. Опытным путем было установлена, что окисление при более высокой постоянной температуре (125°С), хотя и значительно сокращает время реакции, но отрицательно сказывается на качестве синтетических жирных кислот. Процесс окисления прерывается при достижении кислотного число 70 [1].
Практическое осуществление окисления парафинов связано с использованием в этой реакции перманганата калия в качестве катализатора. Изучение непосредственного взаимодействия КМnO4 и MnO2 с парафином затрудняется тем, что эти катализаторы на начальных стадиях реакции находятся в гетерогенном состоянии.
Экспериментальное изучения поведения KMnO4 и MnO2 в среде расплавленного парафина показало, что ни то, ни другое соединение без кислорода не взаимодействует с углеводородами [3].
Это свидетельствует о том, что реакция взаимодействий Мn+2 с радикалами RO2• конкурирует с реакцией продолжения цепи таким образом, что при уменьшении концентрации углеводорода до определенного значения наблюдается полное прекращение процесса окисления.
Введение солей калия стабилизирует марганцовый катализатор и предотвращает выпадение осадка. Более того, добавление стеарата калия к осадку соединений марганца вызывает растворение последнего. Одновременно с этим изменяется и кинетика окисления, увеличивается скорость образования свободных кислот, снижается содержание карбонильных соединений в оксидате [32].
В результате анализа литературы видно, что процессы окисления углеводородов проводятся с участием солей металлов переменной валентности, которые улучшает условия образования кислот.
Литературы
1.Каримов И.А., Мировой финансово – экономический кризис, пути и меры его преодолению в условиях Узбекистана / И.А.Каримов Т.: 2009.- 56 с.
2. Регламент производство синтетических жирных кислот / Волгаградский НПЗ. Волгоград, 1982 – 120с.
3. Эммануэль Н.И. Цепные реакции окисления углеводородов в жидкой фазе / Н.И. Эммануэль – М.: Наука, 1965 – 362 с.
4. Юкельсон И.И., Технология основного органического синтеза / И.И. Юкельсон. М.: Химия, 1968. – 672 с.
5.Эвери Г. Основы кинетики и механизма химической ркакций/ Г.Эвери; пер.с анг. В.В.Смирного – М.: Мир 1978 – 216 с.
6. Денисов Е.Т. Химическая кинетика / О.М. Саркисов, Г.И. Лихтенштейн . – М.: Химия 2000 – 568с.
7. Шипаева Т.А. Синтез и изучение свойств многофункциональных добавок на основе хлорпарафинов: дис.канд.хим.наук/ Шипаева Татьяна Александроовна. – Волгоград – 1998.-120 с.
8. Теоритическое изучение механизма окисления углеводородров молекуляном кислородом / Е.В. Николаева, А.Г. Шамов, Г.М. Хропковский и др.// Нефтехимия – 99; тез. докл V конф. по интенсификации нефтехимических процессов – Нижнекамск – 1999. – с. 103- 105.
9.Общая органическая химия : в 8 т. Т.1 / Д.Бартон, У.Д. Оллис; пер. с англ. С.В. Яроцкого; под ред. Н.К.Когеткова, А.И. Усова – М.: Химия 1982 – 856 с.
10. Oxidation of alkanes b TBHR in the presence of soluble titanum complexes / Fujewata Mashario, Xu Qiang, Souma Yoshie etc.// J.Hol.Catal. – 1999, - №1- P.77-84.
11. Meunier B.S.Oxidation catalysis: Pap. first international conference on porphyrins and phthalocyanines (ICPP-1)/ B.S. Meunier// Porphyrins and phthalocyanines -2000. -№4-P. 353
12. Пат. 6037507 США, МПК С 07 С 29/50. Oxidation process of branched aliphatic hydrocarbons and process for producing the oxide / Nakano Tatsuya, Isliii Yasutalca; заявитель и патентообладатель Daicel Chemical Ind. -№09/037703; заявл. 10.03.98; опубл. 14.03.00. ~3с.
13. Selective oxidation of n-butane on a V-P-O-catalyst: Improvement of the catalytic performance under fuel- rich condition by doping / S.Mota, J.C.Volla, G.Vorbeck. etc. //J.Chem. Soc- 2000.- 2.-P.319-329.
14. Catalytic Shilov chemistri: Plaiimnn chloride- catalyzed oxidation of terminal mctihyl groups by dioxigen /Lin Minren, Slien Chengyu, Garsia- Zayas Eduardo A. etc. // J. Amer.Chem, Soc.-2001. -№5. –P. 1000 -100L.
15. Laszio, J. Csanyl. Investigation of the catalytic behavior of ion-pair complexes of vanadium (5+) in the liquid- phase oxidation of hydrocarbons with molecular Oi / Csanyl Laszio J.Jaky Katoly, Galkaes Gabor // J.Mol. Catal. -2000, -№1-2. –P. 109-124.
16. Артемов, А.В. Новые высокоэффективные катализаторы жидкофазных окислительных процесс >> / А.В. Артемов // Катализ и промышленност. -2000.-№2.–С.18-23.
17. Заявка 19924533 Германия, МПК С 07С 57/07. Verfagen zur Marstcllung von Acrykaurc / Sclnfider Jiirdc, Ncstlcr Gerhard, Miiller- Enge J Klaus Joachim; заявитель и патентообладатель BASF AG.- №19924533.9; заявл. 28. 05. 99; Опубл. 30,11 02.-2с.
18. Заявка 19941315 Германия, МПК С 07 С 407/00. Selective oxidation von kohlenwasscretoffen / Langer Rein hard, Fengler Gerd; заявитель и иатентообладатель BASF AG.- №19941315.0, заявл. 31.08.99; опубл. 01.03.01. -3с.
19. Пат. 5981420 США, МПК В 01 J 31/00. Oxidation catalytic system and oxidation process / Nakano T. Isitt Y; заявитель и потентообладатель Daicel Chemical Ind. Ltd.; Yasutaka Isitt.-№09/024514; заявл. 17.02.98; опубл. 09.11.99.-с.
20. Заявка 19823088 Германия, МПК С 07 С 51/21. Verfahren zur HcistcUung Von Sauren / Riidinger Ch, Eberle H,-J., Bogner R., Kohlmarm W.; заявитель Consortium fur elektrocliemische Industrie GmbH.-№19823088.5; заявл. 22.05.98; опубл. 25.11.99.-4c.
21. Заявка 981066/04 Россия, МТЖ С07 В 41/08. Способ окисления углеводородов, спиртов и /или кетонов / Константины Мишель, Фаш Эрик, Родья Фибер Э.; заявитель Резон Эяермедиа, -№98106628/04; заявл. 09.04.98; опубл. 27.01.00.-6c.
22. Пат. 5914013 США, МПК С 07 В 33/00. Selective Thermal and fotooxidatkm of hydrocarbons in zeolites by oxygen / Frei Hein?., Blatter Fritz, Sun Hai; заявитель и патентообладатель. The Regents of the University of Colifomia.-№08/874,679; заявл. 13.06.97; опубл, 22.06.98.-fic.
23. Заявка 19746667 Германия, МПК С 07 С57/05. Verfahren den heterogen katalysierten Gasphascnoxidation von Propan zu Acrolein nnd/ odcr Acriylsaure / Jachow II , Tsnten A, Univerricht S, Arnold A; заявитель BASF AG.-№19746667.2, заявл. 23.01.99.-7c.
24. Kiyoshi, Otsuka. Селективное окисление с участием оксидов азота/ Otsuka Kiyoshi, Yamanaka Ichiro Shokubai // Catalysis and Catalysis,-1999,-№8. –P.606-612.
25. Farinas, E.T. Pliotoinduced oxidation of hydrocarbons with cobalt (111)- alkylperoxy completes / E.T. Farinas, C.V. Nguyen, F.K. Mascherak // Inorg. Chin. Actc. -1997.-Vol.263, 1-2,P. 17-21.
26. Светланов, КВ, Окисление алканов до карбоновых кислот / Н.В, Светланов, Е.А Николаева //Научная сессия : аннотац. сообщ./ 1СГТУ.-Казань, 2003.-с.34.
27. Пат, 6340420 США, МГЖ В 01 D 61/44. Methods of treating the oxidation mixture of hydrocarbons to respective dibasic acids / Dassel Mark W, Vassiliou Euslathics; заявитель и патентообладатель 09/3458S0; заявл. 30.06.99; опубл. 22.01 т.-Лс.
28. Шт. 6515146 США, МШС С 07 D 307/60, C 07 C 51/16. Process for catalytic selective oxidation of hydrocarbon substrate / Perrcgaard Jens Santamaria Jesus, Menendes Miguel etc; заявитель и патентообладатель Mai dor Topsoe A/S, University of Zaraoza, Du Pout Iberia S.A.-fe 09/654299; заявл. 01.09.00", опубл. 04.02.03.-бс.
29. Заявка 282S194 Франция, МПК С 07 С 51/13, Procede d'oxydation d'hydrocarbures en acides / Bonnet Didicr, Fache Brie, Simotiato Jean Pierre; заявитель P.Jiodia Polyamide Intermediates SAS.-N 0110427; заявл. 03.08.01; опубл. 07.02.03.-4с.
30. Shulpin, G.B. Melall- catalysed hydrocarbon oxygenations in solution: The dramatic role of additives: A review / G.B. Shulpin // J.Mol Calal.A, -2002,-№1.-P.39-66.
31. Заявка 10201241 Германия, МПК В 01 J 31/02. Katalysator / Weisbeck Markus, Mcincii Maric- Therese, Schmirt Jurg etc.; заявитель Bayer AG. -№ 10201241.5; заявл. 15.01.02; опубл. 24.07.D3.-6c.
32. Заявка 2824322 Франция, МГЖ С 07 С 037/00. Precede d'oxydation d'hydrocarbures / Fache Eric, Simonato Jean Prierre (RHODIA SERVICES ); зритель RHODIA POLYAMIDE INTERMEDIATES SAS.- 0106016; заявл. 04.05.01; опубл. 08.11.02.-6с.
33. Заявка 200110090/04 Россия, МПК С 07 С 51/43. Способ выделения и очистки карбоновой кислоты, образующиеся при реакции прямого окисления углеводородов / Константин и Мишель, Фаш Эрик, Маремм Шильбстр; заявитель Родиа Полиамид Интермедиа. – № 200110090/04; заявл.14.04. 99, опубл. 20.01.03.- 4 с.
34. Заявка 19622331 Германия, МПК С 07 С 47/22. Verfahren der Heterogen katalisierten Gaspasenoxidation von Propan zu Acrolein / Tenten A., Proll Th., Schildberg M.-P.; заявитель BASF AG. -№ 19622331.8; заявл. 04.06.96; опубл. 11.12.97.- 4 с.
35. Пат. 5536875 США, МПК С 07 С 51/16. Enhanced oxidation of organic chemicals / Roby Anne K., Kingsley Jeffrey P.; заявитель и пантенто- обладатель Praxair Technology Inc.; заявл. 22.05.95; опубл. 16.07.96.- 3с.
36. Заявка 2732678 Франция, МКИ С 07 С 55/14, С 07 С 51/215. Procede d’oxydation d’hydrocarbures, d’alcogols ou de cetones par catalyse heterogene / Custantini M., Fashe E., Gilbert L.; заявитель Rhone-Paulene Chimie. - №9504428; заявл. 07.04.95; опубл. 11.10.96. – 3 с.
© 2009 База Рефератов